IEPT20131223
Rb. Den Haag, 06-07-2016, nr. C/09/482194 / HA ZA 15-150
Uitspraak 06‑07‑2016
Inhoudsindicatie
IE, octrooirecht, EP NL deel voor rivastigmine pleisters niet inventief en dus niet geldig, ongeldigheid uitsluitend als verweer gevoerd, inbreukvorderingen afgewezen. Proceskosten 1019h Rv
Partij(en)
vonnis
RECHTBANK DEN HAAG
Team handel
Zittingsplaats Den Haag
zaaknummer / rolnummer: C/09/482194 / HA ZA 15-150
Vonnis van 6 juli 2016
in de zaak van
de rechtspersoon naar vreemd recht
NOVARTIS AG,
gevestigd te Basel, Zwitserland,
eiseres,
advocaat mr. D. Knottenbelt te Rotterdam,
tegen
1. de rechtspersoon naar vreemd recht
ALVOGEN IPCO S.a.r.l,
gevestigd te Senningerberg, Luxemburg,
2. de besloten vennootschap met beperkte aansprakelijkheid
FOCUS FARMA B.V.,
gevestigd te Koog aan de Zaan,
gedaagden,
advocaat mr. M.H.J. van den Horst te Den Haag.
Partijen zullen hierna Novartis en (gedaagden gezamenlijk in enkelvoud) Alvogen c.s. genoemd worden en gedaagden ieder afzonderlijk Alvogen en Focus Farma. Voor Novartis is de zaak inhoudelijk behandeld door mr. R.M. Kleemans en mr. W. de Jong, beiden advocaat te Amsterdam met bijstand van drs. K.M.L. Bijvank, octrooigemachtigde. Voor Alvogen c.s. is de zaak behandeld door mr. Van den Horst voornoemd en mr. C. Zeri, advocaat te Den Haag met bijstand van dr. R.E. Raggers, octrooigemachtigde.
1. De procedure
1.1.
Het verloop van de procedure blijkt uit:
- de beschikking van de voorzieningenrechter van deze rechtbank van 7 oktober 2014
waarbij Novartis verlof is verleend om te procederen volgens het Versneld Regime in Octrooizaken;
- -
de exploten van dagvaarding van 4 november 2014 met producties 1-23;
- -
de conclusie van antwoord van 15 april 2015, met producties 1-29;
- -
de akte houdende reactie op bij conclusie van antwoord gebezigde nietigheidsargumenten
van Novartis van 10 juni 2015, met producties 24-28;
- -
de akte houdende overlegging productie 29 van Novartis;
- -
de akte overlegging nadere producties 30-32 van Alvogen c.s.;
- -
de akte houdende overlegging reactieve producties 30-33 van Novartis;
- -
de akte houdende overlegging reactieve productie 34 van Novartis;
- -
de akte houdende overlegging proceskostenoverzicht met productie 35 van Novartis;
- -
de akte overlegging proceskostenoverzicht (productie 33) van Alvogen c.s.;
- -
het aanvullende proceskostenoverzicht van Novartis;
- -
het geactualiseerde proceskostenoverzicht (productie 33.1) van Alvogen c.s.;
- -
het pleidooi van 18 september 2015 en de daarbij door beide partijen overgelegde
pleitnotities, met in de pleitnotities van mr. Kleemans en mr. De Jong doorgehaald de paragrafen 5, 6, 18 t/m 23, 53, 55, 58 t/m 64, 69 welke niet zijn gepleit en in de pleitnotities van mr. Van den Horst en mr. Zeri doorgehaald de paragrafen 142 (vanaf “Deze kennis…”) t/m 155 welke niet zijn gepleit.
1.2.
Ten slotte is vonnis nader bepaald op heden.
2. De feiten
2.1.
Novartis is een wereldwijd opererend farmaceutisch bedrijf.
2.2.
Novartis is een van de houdsters van het Europese octrooi 2 292 219 B1 (hierna: EP 219 of het octrooi) voor een "Transdermal therapeutic system for the administration of rivastigmine", verleend op een aanvrage van 10 oktober 2006, met een beroep op het prioriteitsdocument US 741511 P en de prioriteitsdatum 1 december 2005. Een “transdermal therapeutic system” (hierna ook: TTS) is een medicinale pleister voor transdermaal gebruik. Conclusie 1 zoals aangevraagd is gedurende de verleningsprocedure gewijzigd en uiteindelijk verleend als hierna opgenomen. EP 219 is – onder meer in Nederland – van kracht met ingang van 12 juni 2013. EP 219 expireert op 9 oktober 2026. Tegen de verlening van EP 219 is door verschillende partijen oppositie ingesteld. Ten tijde van het pleidooi is door partijen medegedeeld dat de oral hearings bij de oppositie afdeling van het Europees Octrooibureau (hierna: EOB) zouden plaatsvinden op 15 december 2015. De rechtbank is er ambtshalve mee bekend dat de oppositie afdeling het octrooi wegens toegevoegde materie heeft herroepen. Tegen die beslissing is beroep ingesteld.
2.3.
De (oorspronkelijke) Engelse tekst van conclusie 1 van EP 219 - de enige conclusie van het octrooi als verleend - luidt als volgt:
1. Rivastigmine for use in a method of preventing, treating or delaying progression of dementia or Alzheimer's disease, wherein the rivastigmine is administered in a TTS and the starting dose is that of a bilayer TTS of 5 cm2 with a loaded dose of 9mg rivastigmine, wherein one layer:
has a weight per unit area of 60 g/m2 and the following composition:
- rivastigmine free base 30.0 wt %
- Durotak® 387-2353(polyacrylate adhesive) 49.9 wt %
- Plastoid® B (acrylate copolymer) 20.0 wt %
- Vitamin E 0.1 wt %
and wherein said layer is provided with a silicone adhesive layer having a weight per unit area of 30 g/m2 according to the following composition:
- Bio-PSA® Q7-4302 (silicone adhesive) 98.9 wt %
- Silicone oil 1.0. wt %
- Vitamin E 0.1. wt %
2.4.
De (onbestreden) Nederlandse vertaling van conclusie 1 van EP 219 luidt:
1. Rivastigmine voor toepassing in een werkwijze voor het voorkomen, behandelen of vertragen van de progressie van dementie of de ziekte van Alzheimer, waarbij rivastigmine wordt toegediend in een TTS en de aanvangsdosis die is van een dubbellaag-TTS van 5 cm2 met een geladen dosis van 9 mg rivastigmine, waarbij één laag:
een gewicht per oppervlakte-eenheid van 60 g/m2 en de volgende samenstelling heeft:
- -
rivastigmine, vrije base 30,0 gew.%
- -
Durotak® 387-2353 (polyacrylaat-hechtmiddel) 49,9 gew.%
- -
Plastoid® B (acrylaat-copolymeer) 20,0 gew.%
- -
Vitamine E 0,1 gew.%
en waarbij de genoemde laag is voorzien van een silicone-hechtlaag met een gewicht per oppervlakteeenheid van 30 g/m2 volgens de volgende samenstelling:
- -
Bio-PSA® Q7-4302 (silicone-hechtmiddel) 98,9 gew.%
- -
Siliconenolie 1,0 gew.%
- -
Vitamine E 0,1 gew.%.
2.5.
In de beschrijving van EP 219 zijn onder meer de volgende passages opgenomen:
[0001] The present invention relates to rivastigmine, in free base or pharmaceutically acceptable salt form, for use in a method of preventing, treating or delaying progression of dementia or Alzheimer’s disease, wherein the rivastigmine is administered in a Transdermal Therapeutic System and the starting dose is as defined in claim 1.
[0002] Transdermal Therapeutic Systems (TTS) and their manufacture are generally known in the art. EP 1 047 409 discloses a TTS containing rivastigmine and an antioxidant. GB 2203040 discloses a TTS containing rivastigmine and a hydrophilic polymer.
[0003] These TTS have valuable properties. However, there is a need for further TTS showing improved properties, in particular, there is a need to provide TTS to improve compliance, adhesion, tolerability and/or safety.
(…)
[0006] Thus, it is an aim of the present invention to provide a TTS with improved compliance, adhesion, tolerability and/or safety properties.
[0007] It is a further objective of the present invention to provide a TTS that has a relatively large amount of active ingredient and has an adhesive force to ensure safe application over the entire application period.
[0008] It is a further objective of the present invention to provide a TTS that has a relatively large amount of active ingredient without having an inadequately large expanse.
[0009] It is a further objective of the present invention to provide a TTS that shows improved adhesive properties without changing the release profile of the active ingredient.
[0010] It is a further objective of the present invention to provide a method of treatment and controlled-release formulation(s) that substantially improves the efficacy and tolerability of rivastigmine.
[0011] It is a further objective of the present invention to provide a method of treatment and controlled-release formulation(s) that substantially reduces the time and resources needed to administer rivastigmine for therapeutic benefit.
[0012] It is a further objective of the present invention to provide a method of treatment and
controlled-release formulation(s) that substantially improves compliance with rivastigmine
therapy.
[0013] It is a further objective of the present invention to provide a method of treatment and
controlled-release formulation(s) that have substantially less inter-individual variation with
regard to plasma concentrations of rivastigmine required to produce a therapeutic benefit
without unacceptable side effects.
[0014] This is achieved by a TTS as defined in the enclosed claims.
(…)
[0017] Tests with active ingredients for the treatment of Alzheimer's disease have surprisingly shown that a line of silicone adhesive can be applied to a poorly adhesive reservoir matrix, thus significantly increasing the adhesive properties of the preparation without affecting the thermodynamic properties of the TTS, i.e. without reducing the release of active ingredient from the matrix and its permeation through the skin.
[0018] The findings of the tests on transdermal application of active ingredients for the treatment of Alzheimer's disease carried out by the applicant can of course be transferred to other groups of active ingredients. It can therefore be stated in general that for many active ingredients an increasing proportion of active ingredient in the adhesive polymer matrix of the TTS significantly reduces the adhesive properties of the TTS if said active ingredients are solid at room temperature. Usually, if the active ingredients are in a liquid state at room temperature large amounts of so-called "thickening polymers" (e.g. cellulose or polyacrylate derivatives) have to be added in order to achieve mechanical processability of the polymers, which results also in a reduction of adhesive properties.
[0019] In one embodiment the present disclosure provides TTS comprising a backing layer, a reservoir layer containing at least one active ingredient and a polymer, an adhesive layer comprising a silicone polymer and a tackifier.
[0020] A TTS according to the disclosure shows improved adhesive properties. Further, and very surprisingly, the so obtained TTS has essentially the same release profile when compared with a standard TTS.
[0021] The present invention is further related to substantially improving the efficacy and tolerability of rivastigmine, by application of a TTS in the range of 2 to 50 cm2, said formulation providing a mean maximum plasma concentration of about 1 to 30 ng/mL from a mean of about 2 to 16 hours after application and an AUC24h of about 25 to 450 ng.h/mL after repeated “QD” (i.e., once daily) administration.
[0022] A TTS as used in the invention quite surprisingly shows improved tolerability, particularly gastrointestinal adverse events such as nausea and vomiting, relative to equivalent levels of exposure (AUC24h) of Exelon® capsule.
(…)
[0044] In a further aspect, the invention provides rivastigmine in free base or pharmaceutically acceptable salt form, for use in the prevention, treatment or delay of progression of dementia wherein the rivastigmine is administered in a TTS and the stering is as defined in claim 1.
[0045] In a further aspect, the invention provides rivastigmine for use in a method for the prevention, treatment or delay of progression of dementia associated with Parkinson's disease wherein the rivastigmine is administered in a TTS and the stering is as defined in claim 1.
[0046] In a further aspect, the invention provides rivastigmine for use in a method for the prevention, treatment or delay of progression of Alzheimer's disease wherein the rivastigmine is administered in a TTS and the stering is as defined in claim 1.
[0047] The manufacturing of a TTS according to the invention may be accomplished in any method known to the skilled person.
(…)
[0049] Little has been published in detail on rivastigmine’s biopharmaceutical properties in humans. It is rapidly and completely absorbed. We have found that it is metabolised mainly through hydrolysis by esterases, e.g., acetyl and butyryl cholinesterase and has a plasma half life of 1 hour. It is subject to pre-systemic and systemic metabolism. We now have found that a TTS containing rivastigmine may be produced with advantageous properties, e.g., better tolerability.
[0050] A person skilled in the art is familiar how to produce a TTS having the above defined plasma profiles. A person skilled in the art will appreciate that such plasma profiles may be obtained by varying, e.g.,:
- -
the composition of the first and/or second components, e.g., the nature and amount of excipients and/or active agent(s)
- -
the type of the adhesive layer
- -
the dimension of the patch
[0051] A TTS may be formulated with following aspects in mind:
- -
the time until the release of active agent (lag time or delay time)
- -
the rate of release of active agent (fast or slow)
- -
the duration of release of active agent (long or short)
- -
Reducing first-pass metabolism
- -
Improve compliance of the patients
- -
Reduce application intervals
(…)
[0055] The exact amounts of active agent doses and of the TTS to be administered depend on a number of factors, e.g., the condition to be treated, the desired duration of treatment and the rate of release of active agent.
[0056] For example, the amount of the active agent required and the release rate may be determined on the basis of known in vitro or in vivo techniques, determining how long a particular active agent concentration in the blood plasma remains at an acceptable level for a therapeutic effect.
[0057] The TTS used in the invention allows, e.g., the manufacture of once a day pharmaceutical forms for patients who have to take more than one dose of an active agent per day, e.g., at specific times, so that their treatment is simplified. With such compositions tolerability of rivastigmine may be improved, and this may allow a higher starting dose and a reduced number of dose titration steps.
[0058] An increased tolerability of rivastigmine provided by the compositions may be observed in standard animal tests and in clinical trials.
(…)
IV. Pharmacokinetic properties
[0073] An open-label, parallel-group, four-period, ascending dose-proportionality study evaluating TTS#2 5 cm2, 10 cm2, 15 cm2, and 20 cm2 and 1.5 mg, 3 mg, 4.5 mg, and 6 mg BID [bis in diem, rb.] Exelon® capsules at steady state in patients with mild-to-moderate Alzheimer's disease was conducted.
[0074] Patients diagnosed with mild to moderate Alzheimer's Disease were randomized to either TTS#2 or capsule treatment. The criteria for inclusion were: male or female (non-child-bearing potential) patients, 50-85 years of age, who fulfill the (DSM-IV) criteria for dementia of the Alzheimer's type. Patients should have been diagnosed with probable AD according to NINCDS - ADRDA criteria, with a MMSE score of 10-26 (both inclusive), and no other medical conditions that could impact study results.
[0075] Based on previous experience in clinical trials, 14-day titration steps were implemented for this study.
[0076] At the time of this analysis, the following number of patients completed each of the four periods, and were included in the pharmacokinetic evaluation:
Capsule | TTS#2 |
|---|---|
19 patients in the 1.5 mg bid dose | 18 patients in the 5 cm2 dose |
18 patients in the 3.0 bid dose | 18 patients in the 10 cm2 dose |
13 patients in the 4.5 mg bid dose | 16 patients in the 15 cm2 dose |
12 patients in the 6.0 mg bid dose | 11 patients in the 20 cm2 dose |
[0077] The pharmacokinetics of rivastigmine were investigated after both treatments on the last day of each titration period, except on highest doses when it is investigated on third day of titration (in order not to miss plasma samplings in case of early drop-outs due to poorer tolerability). Plasma samples were analyzed for rivastigmine using LC-MS/MS with a lower limit of quantification (LLOQ) of 0.2 ng/mL. Standard noncompartmental pharmacokinetic parameters were derived from the individual plasma concentration-time profiles using WinNonlin Pro.
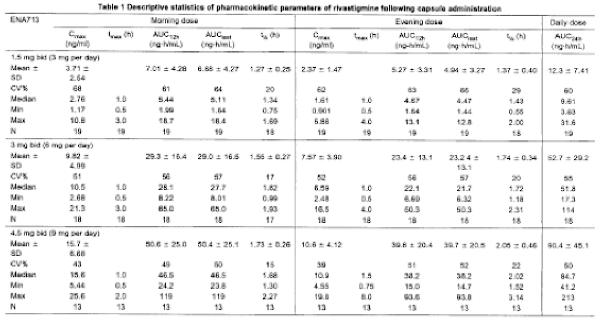
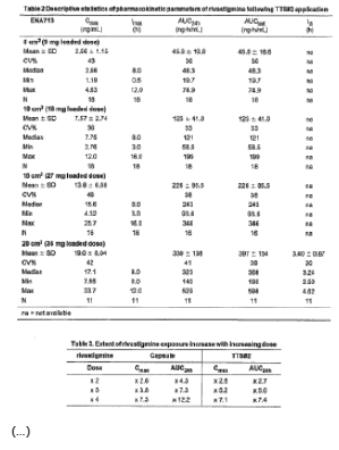
[0078] The pharmacokinetic parameters of rivastigmine are summarized in Table 1 (capsule treatment) and Table 2 (TTS#2 treatment). The mean (± SD) plasma concentration-time profiles are displayed in Figure 4.
[0079] During the application of TTS#2, a rivastigmine plateau concentration was achieved at a median tmax of 8.0 h for all TTS sizes. Exposure also increased over-proportionally with increasing doses as displayed in Table 3, but to a lesser extent than with the capsule, in particular for AUC24h.
[0080] The inter-subject variability as assessed by the coefficients of variation (CVs) for the exposure parameters of rivastigmine (Cmax and AUC24h) was generally lower after the patch (CVs of 33-48%) as compared to the oral administration (CVs of 39-68%).
V. Pharmacologic properties
[0081] TTS#2 shows improved pharmacological properties when compared with a capsule formulation as shown in standard animal test and in clinical trials.

(…)
2.6.
Vóór de prioriteitsdatum van het octrooi bracht Novartis rivastigmine op de markt in de vorm van een tweemaal daags in te nemen capsule met een (niet therapeutisch effectieve) aanvangsdosis van 1,5 mg rivastigmine. Deze capsules voor orale toediening worden door Novartis op de markt gebracht onder de merknaam Exelon®.
2.7.
In de samenvatting van productkenmerken van de orale Exelon capsules (hierna: de Exelon SmPC) staat het volgende doseringsregime:
4. KLINISCHE GEGEVENS
4.1
Therapeutische indicaties
Symptomatische behandeling van milde tot matig ernstige dementie van het Alzheimer-type.
4.2
Dosering en wijze van toediening
Toediening: De start van het toezicht op de behandeling dient te geschieden door een arts met
ervaring in de diagnose en behandeling van dementie van het Alzheimer-type. De diagnose dient
gesteld te worden aan de hand van de huidige richtlijnen. De behandeling met rivastigmine mag
slechts gestart worden, indien er een verzorger beschikbaar is, die regelmatig de geneesmiddeleninname door de patiënt bewaakt.
Rivastigmine dient tweemaal daags te worden toegediend, bij het ontbijt en de avondmaaltijd. De
capsules dienen in hun geheel doorgeslikt te worden.
Startdosis: 1,5 mg tweemaal daags.
Dosistitratie: De startdosis is tweemaal daags 1,5 mg. Wanneer deze dosering na minimaal twee
weken behandeling goed verdragen wordt, kan de dosis verhoogd worden tot tweemaal daags 3 mg.
Verdere verhogingen tot 4,5 mg en vervolgens 6 mg tweemaal daags zijn mede afhankelijk van het
goed verdragen worden van de huidige dosis en kunnen worden overwogen na minimaal twee weken
behandeling bij die dosis.
Wanneer bijwerkingen (b.v. misselijkheid, braken, buikpijn, of verlies van eetlust) of een gewichtsafname tijdens de behandeling worden waargenomen, kunnen deze reageren op het overslaan van één of meerdere toedieningen. Wanneer de bijwerkingen aanhouden, dient de dagelijkse dosis tijdelijk verminderd te worden tot de voorheen goed verdragen dosis.
Onderhoudsdosis: De effectieve dosis is tweemaal daags 3 tot 6 mg. Om een maximaal therapeutisch effect te bereiken dienen patiënten te worden ingesteld op de hoogste door hen goed verdragen dosis.
De aanbevolen maximale dagelijkse dosis is tweemaal daags 6 mg.
De onderhoudsbehandeling kan voortgezet worden zo lang er een therapeutisch voordeel voor de
patiënt bestaat. Daarom dient het klinisch voordeel van rivastigmine regelmatig opnieuw geëvalueerd te worden, met name bij patiënten die behandeld worden met doseringen lager dan tweemaal daags 3 mg. Indien er geen bewijs meer aanwezig is van een therapeutisch effect dient het staken van de therapie overwogen te worden. De individuele respons op rivastigmine kan niet voorspeld worden. Het effect van de behandeling is niet onderzocht in placebo-gecontroleerde studies, die langer duurden dan 6 maanden.
2.8.
Sinds 2007 brengt Novartis pleisters met rivastigmine op de markt. Het gaat om een pleister met een geladen dosis van 9 mg met dosering actief bestanddeel van 4,6 mg per 24 uur in een pleister van 5 cm², een pleister met een geladen dosis van 18 mg en een dosering van 9,5 mg per 24 uur in een pleister van 10 cm² en een pleister met een dosering van 13,3 mg per 24 uur (waarvan de geladen dosis en de maat niet is vermeld in de processtukken). De pleisters worden eveneens onder de merknaam Exelon verhandeld (hierna: de Exelon pleisters). Volgens Novartis vallen de Exelon pleisters onder de beschermingsomvang van het octrooi.
Alvogen c.s.
2.9.
Op 12 maart 2013 heeft Alvogen van het College ter Beoordeling van Geneesmiddelen marktvergunningen verkregen voor Nederland met betrekking tot de producten “Permente 4,6 mg/24hr patch for transdermal use” en “Permente 9,5 mg/24hr patch for transdermal use” (hierna ook: de Permente pleisters). Focus Farma is leverancier van de (door een derde in Zuid-Korea vervaardigde) Permente pleisters. De Permente pleisters zijn opgenomen in de zogenaamde G-standaard waarbij Focus Farma staat genoemd als productverantwoordelijke en Alvogen als registratiehouder.
2.10.
Novartis heeft een kort geding tegen Alvogen c.s. gevoerd dat resulteerde in een vonnis van de voorzieningenrechter van deze rechtbank van 23 december 2013.De voorzieningenrechter oordeelde dat er een serieuze niet te verwaarlozen kans bestaat dat EP 219 zal worden herroepen c.q. vernietigd wegens toegevoegde materie en wees de vorderingen af. Het Gerechtshof Den Haag heeft dit vonnis in zijn arrest van 18 november 2014 bekrachtigd.
Stand van de techniek
2.11.
Uit de stand van de techniek doet Alvogen c.s. een beroep op de Amerikaanse octrooiaanvrage US 2001/0048938 (hierna: US 938) voor een “TTS containing an antioxidant”, aangevraagd op 20 december 2000 en gepubliceerd op 6 december 2001.
In US 938 staat voor zover relevant het volgende.
[0001] This invention relates to a pharmaceutical composition for systemic administration of a phenyl carbamate, e.g. by transdermal administration. In particular this invention relates to a pharmaceutical composition of the phenyl carbamate—(S)-N -ethyl-3-[1-dimethylamino)ethyl]-N-methyl-phenyl-carbamate—(hereinafter referred to as compound A) in free base or acid addition salt form as disclosed in published UK patent application GB 2 203 040, the contents of which are incorporated herein by reference.
[0002] Compound A is useful in inhibiting acetylcholinesterase in the central nervous system, e.g. for the treatment of Alzheimer's disease.
(…)
[0011] As a polymer one can mention in particular an acrylate co-polymer, e.g. co-polymers of butyl acrylate, ethyl hexyl acrylate and vinyl acetate. Preferably the polymer is cross-linked. A preferred acrylate polymer is one of the Durotak brand available from National Starch and Chemical Company, Zutphen, Holland. e.g. Durotak 87-2353 (hereinafter polymer A), 387-2051 or 387-2052 (hereinafter polymer D).
(…)
[0016] Examples of commercially available polymers of this type include:
[0017] 1) Polymers of methacrylate containing alkyl (C1-4)ester groups. Preferably the polymer matrix is a mixture of an acrylate polymer and a methacrylate polymer e.g. in a weight ratio of from 5:1 to 1:1, e.g. 4:1 to 2:1 e.g. 3:1, e.g. butylrnethylacrylate and methylmethylacrylate. MW 20000, e.g. Plastoid B from Röhm, Darmstadt, Germany (hereinafter polymer B).
(…)
[0019] 3) Polymers of methacrylate esters containing trimethylaminoethyl cationic ester groups and other neutral (C1-4)alkyl ester groups. Chloride ions may be present. Mean molecular weight 150,000. Viscosity (20º C.) 10 cP. Refractive Index 1.38. Density 0.815.
Alkali number of 180 mg KOH per gram polymer (Eudragit E 100, Registered Trade Mark, also available from Röhm and hereinafter referred to a polymer C).
(…)
[0061] In a further embodiment, the invention provides a transdermal device comprising a backing layer, a layer comprising compound A in a polymer matrix, a release-liner and, disposed between the layer comprising compound A in a polymer matrix and the release liner, a discrete layer of adhesive material for releasably fixing said transdermal device to patients skin.
[0062] Preferably, the adhesive material is a silicone adhesive chosen from amine-resistant silicone pressure sensitive adhesives as hereinabove described.
[0063] Typically, a transdermal device of said further embodiment comprises:
[0064] a) a polymethacrylate backing layer
[0065] b) Compound A in free base form in an acrylate copolymer
[0066] c) a BIO-PSA Q74302 silicone adhesive layer
[0067] d) a release-liner.
[0068] Preferably, said further embodiment also comprises silicone oil, e.g. silicone oil Q7-9120 from Dow Coming Corporation, in an amount of 0.1 to 5% by weight, e.g. 1%. The backing layer thickness is preferably from 10 to 50 μm,, e.g. 23 μm, and has preferably a round shape.
(…)
[0073] It is an optional feature of all the transdermal devices described hereinabove that they comprise a layer of adhesive between the pharmaceutical composition and the release liner. This, has the primary function of fixing the release liner in contact with the remainder of the device thus protecting the pharmaceutical composition before use. However, if the adhesive is a silicone adhesive, then the layer may additionally act as a membrane through which the Compound A may pass at a controlled rate into the patient through the skin. Without wishing to be limited to a particular theory, it is suggested that the Compound A, dispersed throughout the polymer matrix exhibits little tendency to migrate into the silicone adhesive layer during storage. Accordingly, there is relatively low concentration of Compound A in the silicone layer. In use, the subjects skin, however, may display a much higher affinity for Compound A than the silicone layer and the initial low concentration of Compound A in the silicone layer passes into the subject's body. The silicone layer surprisingly prevents the subject from receiving a sudden high dose of Compound A upon
application of the device and instead promotes a gradual increase of concentration in the subject.
(…)
[0075] The transdermal devices of the invention in general have, for example an effective contact area of pharmaceutical composition on the skin of from about 1 to about 80 square centimeters, preferably about 10 square centimeters, and are intended to be applied at intervals of about once every 1 to 7 days, preferably 1-3 days. Compound A is well tolerated at a dose of 36 mg in free base form in up to 80 cm² of patches according to the invention containing 36 mg compound A from which 12 mg was absorbed. Compound A may, for example be administered at a dose of 8 mg in a patch of ca. 10 cm², once every day. The patch may be applied, for example on the abdomen, thigh, behind an ear, or on a shoulder or upper arm.
[0076] The pharmaceutical composition, optionally formed as a transdermal device, of the present invention are useful for the same indications as for known compositions containing compound A. The exact amounts of compound A to be administered may depend on a number of factors, e.g. the drug release characteristics of the compositions, the drug penetration rate observed in vitro and in vivo tests, the duration of action required, the form of compound A, and for transdermal compositions the size of the skin contact area, and the part of the body to which the unit is fixed. The amount of and, e.g. area of the composition etc. may be determined by routine bioavailability tests comparing the blood levels of active agents after administration of compound A in a composition according to the invention to intact skin and blood levels of Compound A observed after oral administration of a therapeutically effective dose of the compound.
[0077] Orally, the Compound A is well tolerated at an initial dose of 1.5 mg twice a day orally and the dose may be stepped up to 3 mg twice daily in week 2. Higher dosages are possible, for example 4.5 mg twice daily and even 6 mg twice daily. Tolerability is seen to be even better for the transdermal device, wherein 24 mg were absorbed in 24 hours.
[0078] The following example illustrates the invention.
(…)
EXAMPLE 4
[0085] A two-parts composition is prepared consisting of the following components
Composition Per Unit (10 cm²)
[0086]
Compound A | 18 | mg | 30% |
Polymer | 29.94 | mg | 49.85% |
methacrylate | 12 | mg | 20% |
α-tocopherol | 0.06 | 0.1% | |
Total 1st part | 70 | mg | 100% |
(area weight 60 mg/10 cm2) | |||
-continued | |||
and | |||
Bio-PSA Q7-4302 | 29.67 | mg | 98.9% |
Silicone oil Q7-9120 | 0.3 | mg | 1.0% |
α-tocopherol | 0.03 | mg | 0.1% |
Total 2nd part | 30 | mg | 100% |
(area weight 30 mg/10 cm2) | |||
[0087] The two parts arc then put together in the form of a patch.
(…)
[Claims, toevoeging rb].
13. A transdermal device comprising a backing layer, a layer comprising (S)-N-ethyl-3-[1-dimethylamino)ethyl]-N-methyl-phenyl-carbamate in a polymer matrix, a release liner
and, disposed between the layer comprising (S)-N-ethyl-3 [l-dimethylamino)ethyl]-N-methyl-phenylcarbamate in a polymer matrix and the release liner, a discrete layer of adhesive material for releasably fixing said transdermal device to patients skin.
Parallelle procedures
2.12.
Ook de buitenlandse delen van EP 219 zijn onderwerp geweest van procedures. In diverse inbreukprocedures (voornamelijk voorlopige voorzieningen) zijn in een aantal landen voorzieningen opgelegd en in andere landen voorzieningen geweigerd dan wel in hoger beroep opgeheven. Daarbij is in de meeste gevallen geen wezenlijke geldigheidstoets uitgevoerd. In de hierna genoemde parallelle bodemprocedure is de geldigheid van EP 219 wel aan de orde geweest.
2.13.
In twee gevoegde inbreukprocedures die Novartis tegen Focus Pharmaceuticals en Actavis en tegen Teva had aangespannen, heeft de Engelse High Court (Arnold J) bij vonnis van 27 april 2015 ([2015] EWHC 1068 (Pat)) geoordeeld dat het Britse deel van EP 219 nietig is vanwege toegevoegde materie en een gebrek aan inventiviteit ten opzichte van het Amerikaanse octrooi US 6,335,031 (hierna: US 031).
3. Het geschil
3.1.
Novartis vordert – samengevat – dat de rechtbank, bij vonnis, voor zover mogelijk uitvoerbaar bij voorraad,
1. Alvogen c.s. gebiedt met onmiddellijke ingang na betekening van het vonnis iedere directe of indirecte inbreuk op (het Nederlandse deel van) EP 219 te staken en gestaakt te houden, meer in het bijzonder het in of voor haar bedrijf te (doen) vervaardigen, te (doen) gebruiken, in het verkeer te (doen) brengen of verder te (doen) verkopen, te (doen) verhuren, af te (doen) leveren en/of anderszins te (doen) verhandelen, dan wel voor een of ander aan te (doen) bieden of in voorraad te (doen) hebben van de Permente pleisters te staken en gestaakt te houden;
2. Alvogen c.s. gebiedt om binnen 30 kalenderdagen opgave te doen van de omzet en bruto winst die met de Permente pleisters gerealiseerd is, gecertificeerd door een onafhankelijk registeraccountant, alsmede opgave te doen van alle overige voor de berekening van de winst en/of schadevergoeding van belang zijnde informatie;
3. Alvogen c.s. gebiedt de geleden schade van Novartis te vergoeden dan wel de door de
octrooi-inbreuk genoten winst aan Novartis af te dragen een en ander naar keuze van Novartis en nader op te maken bij staat;
4. Alvogen c.s. gebiedt binnen een periode van 7 dagen al haar afnemers, voor zover deze afnemers gevestigd zijn in Nederland, schriftelijk te verzoeken de Permente pleisters te retourneren met het aanbod de factuurprijs en transportkosten te vergoeden, met gebruikmaking van de tekst opgenomen in de dagvaarding, onder gelijktijdige toezending van kopieën van deze brief alsmede een lijst van geadresseerden met volledige adresgegevens aan de raadslieden van Novartis;
5. Alvogen c.s. gebiedt tot het doen verwijderen van de Permente pleisters uit deG-standaard;
6. Alvogen c.s. gebiedt aan Novartis een onmiddellijk opeisbare dwangsom te betalen van € 100.000,- voor elke dag of gedeelte daarvan of, zulks ter keuze van Novartis, per Permente pleister, waarop het aan Alvogen c.s. kan worden toegerekend dat de geboden in het vonnis niet geheel of niet deugdelijk worden nageleefd, waarbij elk aangetroffen exemplaar van de Permente pleisters geldt als een afzonderlijke overtreding;
7. Alvogen c.s. veroordeelt in de kosten van dit geding conform artikel 1019h van het Wetboek van Burgerlijke Rechtsvordering (hierna: Rv), te vermeerderen met de kosten door Novartis gemaakt in de kort geding procedure en de aldaar gevorderde kosten en de aan Alvogen c.s. betaalde bedragen, te vermeerderen met nakosten en wettelijke rente.
3.2.
Novartis heeft hiertoe aangevoerd dat de 5 cm² Exelon pleister de samenstelling heeft van de dubbellaags TTS zoals gedefinieerd in conclusie 1 van EP 219 (hierna ook wel: TTS#2). Omdat de Permente pleisters van Alvogen met rivastigmine als actief bestanddeel volgens Alvogen zelf bioequivalent zijn aan de Exelon pleisters, voldoen de Permente pleisters aan alle kenmerken van conclusie 1 van EP 219. Alvogen en Focus Farma maken dan ook inbreuk op conclusie 1 van (het Nederlandse deel van) EP 219, aldus Novartis.
3.3.
Alvogen c.s. voert gemotiveerd verweer. Zij voert aan (uitsluitend als verweer zonder in reconventie de vernietiging van het Nederlandse deel van EP 219 te vorderen) dat EP 219 ongeldig is en dat zij (ook om die reden) daarop geen inbreuk maakt.
3.4.
Op de stellingen van partijen wordt hierna, voor zover van belang, nader ingegaan.
4. De beoordeling
Bevoegdheid
4.1.
De internationale bevoegdheid van deze rechtbank om van de vorderingen van Novartis kennis te nemen, is mede gelet op het hierna te bespreken nietigheidsverweer en nu de dagvaarding is uitgebracht vóór 10 januari 2015, gebaseerd op artikel 64 EVEXjo. artikel 5 lid 3 jo. artikel 22 lid 4 van Verordening (EG) 44/2001 van de Raad betreffende de rechterlijke bevoegdheden, de erkenning en de tenuitvoerlegging van beslissingen in burgerlijke en handelszaken (hierna: EEX-Vo Oud), respectievelijk op artikel 2 jo. artikel 22 lid 4 EEX-Vo Oud. De relatieve bevoegdheid van de rechtbank volgt uit artikel 80 lid 2 onder a van de Rijksoctrooiwet 1995 (hierna: ROW). Overigens is Alvogen c.s. verschenen zonder de bevoegdheid van de rechtbank te bestrijden.
Schorsing
4.2.
Novartis heeft desgevraagd bij pleidooi aangegeven er de voorkeur aan te geven dat de rechtbank de procedure niet schorst als bedoeld in artikel 83 lid 4 ROW 1995 in afwachting van een definitieve beslissing in de aanhangige oppositieprocedure. Alvogen c.s. heeft aangegeven geen bezwaar te hebben tegen een schorsing. Gelet op het standpunt van Novartis en meewegende dat tegen de beslissing van de oppositie afdeling van het EOB hoger beroep bij de technische kamer van beroep is ingesteld, zodat het nog geruime tijd kan duren alvorens in de oppositieprocedure definitief omtrent de geldigheid is beslist, zal de rechtbank in dit geval geen gebruik maken van haar discretionaire bevoegdheid om de behandeling van de zaak te schorsen.
Korte inleiding op de techniek
4.3.
De volgende beschrijving van de technische achtergrond van EP 219 is ontleend aan de wederzijds niet bestreden uiteenzettingen van partijen en overigens niet in geschil.
4.4.
De ziekte van Alzheimer is een degeneratieve en uiteindelijk fatale aandoening. Er is geen genezing voor de ziekte van Alzheimer. Behandeling is gericht op het verminderen van symptomen en het vertragen van de progressie van de ziekte.
4.5.
Dementie - één van de bekende symptomen van de ziekte van Alzheimer - komt voort uit een gereduceerde activiteit van cholinergische neuronen. Neuronen zijn de cellen in het menselijk zenuwstelsel die informatie tussen cellen sturen en verwerken (via synapsen) door neurotransmitters. Acetylcholine (hierna: ACh) is zo'n neurotransmitter. Degeneratie van cholinergische neuronen lijdt tot een verlies van ACh in de hersenen. Dit raakt aan, onder andere, de cognitieve vaardigheden van een patiënt.
4.6.
Acetylcholine esterase (hierna: AChE) is een enzym dat ACh in inactieve moleculen choline en een acetaat-groep afbreekt. Om de concentratie ACh in de hersenen van patiënten die aan milde tot gematigde dementie als gevolg van de ziekte van Alzheimer of Parkinson lijden te verhogen, kunnen AChE remmers worden toegediend. Bij patiënten met dementie kan hierdoor het geheugenverlies minder ernstig worden. Rivastigmine is zo’n AChE remmer.
4.7.
Rivastigmine is aanwezig in vrije base vorm in de pleister formuleringen (TTS) en in de vorm van het waterstoftartraat-zout in de orale doseringsvormen.
4.8.
Toediening van rivastigmine kan leiden tot behoorlijke bijwerkingen zoals misselijkheid en overgeven.
4.9.
Gelet hierop werd op de prioriteitsdatum bij de behandeling een therapeutisch niet effectieve aanvangsdosis rivastigmine aan de patiënt toegediend. Deze therapeutisch niet effectieve aanvangsdosering had tot doel dat het lichaam van de patiënt zich kon aanpassen aan de medicatie, hetgeen helpt bijwerkingen te voorkomen. De dosis werd vervolgens stapsgewijs verhoogd tot de hoogst getolereerde dosering, welke patiënt afhankelijk is. Als een volgende dosering resulteerde in onaanvaardbare bijwerkingen werd de dosering verlaagd. Dit proces van het geleidelijk aanpassen van de dosering wordt titratie genoemd.
4.10.
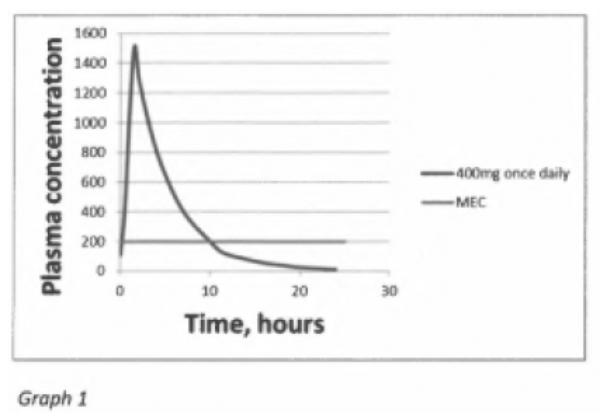
De concentratie werkzame stof in het bloedplasma van een patiënt wordt uitgedrukt in zowel een piekconcentratie in het bloedplasma uitgedrukt in Tmax (de tijd tot de maximale concentratie) en in Cmax (de maximale concentratie op Tmax) als in een hoeveelheid werkzame stof in het bloedplasma van de patiënt gedurende een bepaalde tijdseenheid na toediening. Dit laatste wordt uitgedrukt als een AUC in de eerste x uur (Area Under the Curve in the first x hours). Om een gewenst therapeutisch effect te hebben dient in het algemeen de concentratie van de werkzame stof in het bloedplasma boven een bepaald minimum te liggen (de MEC, minimale effectieve concentratie) en om dat niveau voor langere tijd te behouden zal de Cmax in een onmiddellijke afgifte formulering substantieel boven de MEC moeten liggen. Hierna is ter illustratie een grafiek opgenomen afkomstig uit het rapport van Alvogen’s deskundige Rue (productie GP30). De grafiek laat een geneesmiddel zien met een Tmax van ongeveer 2 uur, een Cmax van ongeveer 1500 mg/ml en een MEC van 200 mg/ml. In het voorbeeld blijft de Cmax gedurende 10 uur boven de MEC liggen. De AUC is het gebied dat ligt onder de curve.
Inventiviteit EP 219
4.11.
Het meest verstrekkende verweer van Alvogen c.s. is dat zij geen inbreuk maakt omdat EP 219 ongeldig is. Zij voert daartoe aan dat EP 219 ongeoorloofde toegevoegde materie bevat, niet nawerkbaar is, niet nieuw is en niet inventief is.
4.12.
Volgens Novartis openbaart EP 219 (behalve een TTS met een siliconen hechtlaag voor verbeterde kleefkracht) een TTS doseringsregime voor het toedienen van rivastigmine voor de behandeling van dementie of de ziekte van Alzheimer aan de hand van een aanvangsdosis. De doelstelling van de uitvinding zoals geopenbaard in het octrooi is volgens Novartis om te voorzien in een behandeling met rivastigmine met een verbeterde therapietrouw en veiligheid terwijl de behandeling door de patiënt ook beter kan worden verdragen. Dit wordt bereikt door middel van het doseringsregime met een inventieve aanvangsdosis zoals gedefinieerd in de enige conclusie van het octrooi waarmee de patiënt met een kortere titratieperiode uitkomt bij de voor hem therapeutisch effectieve dosering, aldus nog steeds Novartis. De aanvangsdosering volgens de uitvinding is al een therapeutisch effectieve dosis.
4.13.
Novartis verdeelt conclusie 1, de enige conclusie van EP 219, in de volgende kenmerken: a) rivastigmine, b) voor toepassing in een werkwijze voor het voorkomen, behandelen of vertragen van de progressie van dementie of de ziekte van Alzheimer, c) waarbij rivastigmine wordt toegediend door een TTS, d) en de aanvangsdosis die is van een referentiepleister. Conclusie 1 ziet volgens Novartis op een aanvangsdosering die bioequivalent is aan de dosering afgegeven door de referentiepleister. De samenstelling van de referentiepleister die is opgenomen in conclusie 1 komt overeen met TTS#2 uit voorbeeld IV van EP 219. Een TTS of pleister met dezelfde aanvangsdosis als de referentiepleister valt onder de beschermingsomvang van conclusie 1 ook als die TTS of pleister een andere structurele samenstelling heeft dan de referentiepleister zoals opgenomen in conclusie 1. Dit komt tot uitdrukking in de tekst van conclusie 1 ‘the starting dose is that of a’, aldus Novartis (hierna ook: de ruime lezing).
4.14.
Conclusie 1 van EP 219 is, uitgaande van de ruime lezing die Novartis bepleit, naar het oordeel van de rechtbank nietig wegens gebrek aan inventiviteit. Die ruime lezing wordt weliswaar door Alvogen c.s. betwist (zij meent dat conclusie 1 uitsluitend een doseringsregime onder bescherming stelt met een aanvangsdosering die wordt afgegeven door een TTS met de samenstelling, maat en dosis zoals beschreven in de conclusie), maar zij heeft haar inventiviteitsaanval wel (tevens) aan de hand daarvan ingericht.
4.15.
Partijen zijn het niet eens over de vraag wat de betekenis is van de term ‘aanvangsdosis’. De rechtbank gaat er in het navolgende veronderstellenderwijs vanuit dat de term ‘aanvangsdosis’ de betekenis heeft die Novartis bepleit, namelijk dat dit de initiële dosis werkzame stof is die wordt toegediend bij de aanvang van de behandeling van de patiënt. Bij een TTS gaat het dan om de initiële dosis werkzame stof die bij aanvang van de behandeling middels de TTS wordt afgegeven aan de patiënt, zo begrijpt de rechtbank. De ‘geladen dosis’ is de hoeveelheid werkzame stof aanwezig in de TTS. De ‘aanvangsdosis’ verschilt van de ‘geladen dosis’ omdat bij een TTS niet alle werkzame stof daadwerkelijk wordt afgegeven aan de patiënt.
4.16.
Dat de uitvinding volgens EP 219 op een voor de hand liggende wijze uit de stand van de techniek voortvloeit, zal hierna aan de hand van de problem solution approach (hierna: PSA) worden toegelicht, welk hulpmiddel ook beide partijen in hun argumentatie hanteren.
4.17.
De rechtbank is met Novartis van oordeel dat de gemiddelde vakman niet slechts een formuleringsdeskundige is, maar dat het gaat om een team bestaande uit een clinicus en een formuleringsdeskundige. De clinicus heeft verstand van farmacokinetiek en behandelt patiënten die lijden aan dementie of de ziekte van Alzheimer. De clinicus is in staat om effectiviteit en bijwerkingen van een therapie voor dementie of de ziekte van Alzheimer, in het bijzonder een doseringsregime, te herkennen. De formuleringsdeskundige is in staat een TTS te maken (vgl. paragraaf 9 pleitnota Novartis).
4.18.
De rechtbank stelt voorop dat als meest nabije stand van de techniek die publicatie wordt genomen die de combinatie van kenmerken toont die het meest geschikte uitgangspunt oplevert (most promising springboard) in de richting van een voor de hand liggende ontwikkeling naar de geclaimde uitvinding. Het moet bij deze selectie gaan om hetzelfde of een nauw verwant technisch gebied en hetzelfde doel als dat van de geclaimde uitvinding. Dat deze meest nabije stand van de techniek op zichzelf met gebruikmaking van kennis van de uitvinding wordt gevonden, is inherent aan de PSA.
4.19.
De rechtbank volgt Alvogen c.s. in haar stelling dat US 938 (zie 2.11) de meest nabije stand van de techniek is. Alvogen c.s. heeft onweersproken gesteld dat transdermale therapeutische systemen met rivastigmine voor gebruik in de behandeling van dementie of de ziekte van Alzheimer al bekend waren vóór de prioriteitsdatum, waaronder in US 938. Voorts heeft Alvogen c.s. onweersproken gesteld dat US 938 leert dat rivastigmine als werkzame stof (‘Compound A’ genoemd - zie par. [0001]) nuttig is voor de behandeling van de ziekte van Alzheimer (par. [0002]), dat transdermaal toegediende rivastigmine goed wordt getolereerd bij een dosering van 36 mg (par. [0075]) en dat de verdraagbaarheid van het actieve ingrediënt door de patiënt bij gebruik van een TTS beter is in vergelijking tot het orale doseringsregime (par. [0077]). In par. [0075] beschrijft US 938 voorts dat de TTS een oppervlakte kan hebben van 1 tot 80 cm² en gedurende 1-7 dagen kan worden gedragen. Ook niet weersproken is de stelling van Alvogen c.s. dat de TTS in US 938 dezelfde structurele samenstelling heeft, inclusief siliconen hechtlaag voor verbeterde kleefkracht, als de TTS#2 zoals gedefinieerd in conclusie 1 van EP 219. Die samenstelling volgt (onder meer) uit voorbeeld 4 van US 938 die ziet op een TTS van 10 cm² met exact dezelfde gewichtsverhoudingen en samenstelling als TTS#2 van 5 cm² uit EP 219. Gelet hierop biedt US 938 naar het oordeel van de rechtbank het meest veelbelovende uitgangspunt om tot de in het octrooi geclaimde aanvangsdosis volgens de referentiepleister met de samenstelling van TTS#2 te komen.
4.20.
De rechtbank verwerpt de stelling van Novartis, die zij inneemt met verwijzing naar haar deskundige prof. dr. Fahr , dat de orale Exelon capsule (zie 2.6) de meest nabije stand van de techniek is. Ook als juist is dat op de prioriteitsdatum een behandeling met orale Exelon capsules de enige vorm van rivastigmine behandeling was waarvoor een handelsvergunning was verleend en waarvan een klinisch beproefde dosering bekend was, is het daarmee nog niet de publicatie met de meeste zelfde kenmerken als EP 219. Behalve dat, zoals hiervoor aangegeven, US 938 een transdermaal therapeutisch systeem openbaart en daarmee de meest veelbelovende springplank voor de vakman is voor een ontwikkeling die zou leiden tot de uitvinding, wordt in par. [0077] van US 938 ook verwezen naar een oraal doseringsregime met een aanvangsdosis van 1,5 mg tweemaal daags dat overeenkomt met het doseringsregime als vermeld in de Exelon SmPC (zie 2.7). US 938 bevat derhalve ook alle technische kenmerken van de door Novartis bepleite meest nabije stand van de techniek.
4.21.
Uitgaande van het in US 938 geopenbaarde transdermale therapeutische systeem als meest nabije stand van de techniek is de verschilmaatregel de aanvangsdosering van EP 219. Het technisch effect van die maatregel is dat de aanvangsdosis al therapeutische werking heeft, maar ook een acceptabel niveau van bijwerkingen. Hierdoor zijn minder titratiestappen nodig om tot de optimale en veilige dosis te komen terwijl de patiënt sneller effect merkt en de behandeling beter volhoudt. In EP 219 staat dit ook als doel van de uitvinding beschreven in paragrafen [0003], [0006], [0021] en [0022].
4.22.
Het objectieve technische probleem dat met de uitvinding wordt opgelost, kan worden geformuleerd als hoe de patiënt versneld in te stellen op voor hem/haar goed te verdragen en veilige dosering voor een behandeling met rivastigmine.
4.23.
Gesteld voor het zojuist geformuleerde probleem verschaft US 938 de gemiddelde vakman naar het oordeel van de rechtbank een aanwijzing (pointer) op grond waarvan hij op de prioriteitsdatum zonder inventieve denkarbeid zou (‘would’ - not ‘could’) komen tot de aanvangsdosering van EP 219.
4.24.
In par. [0076] van US 938 staat dat de keuze voor de juiste maat en dosering voor behandeling met een TTS wordt gemaakt door toepassing van ‘routine bioavailability tests’ waarmee bloedwaarden worden vergeleken op aanwezigheid van rivastigmine na gebruik van enerzijds een TTS volgens US 938 en anderzijds na orale toediening van een therapeutisch effectieve dosis rivastigmine. De vakman wordt dus op het spoor gezet om rivastigmine (i) middels een TTS zoals geopenbaard in US 938 en (ii) uitgaande van een oraal therapeutisch effectieve dosis, toe te dienen en dan door middel van het toepassen van routine tests (van de aanwezigheid van rivastigmine in het bloed) de bloedwaarden te vergelijken om te komen tot een dosering voor een TTS behandeling met rivastigmine. Dat het hierbij inderdaad gaat om routine tests waarmee de geladen dosis en (daarmee) de maat kunnen worden bepaald, is door Novartis niet weersproken.
4.25.
US 938 verschaft de vakman informatie over structuur en samenstelling van een TTS. Zo volgt uit par. [0075] dat een TTS verschillende formaten kan hebben (van 1 tot 80 cm², bij voorkeur 10 cm2). In voorbeeld 4 van US 938 is de samenstelling opgenomen van een TTS van 10 cm² met een geladen dosis van 18 mg. Dat de gemiddelde vakman op de prioriteitsdatum in staat was om TTS-en te maken met eenzelfde samenstelling maar een andere maat en andere geladen dosis is niet in geschil.
4.26.
US 938 verschaft de vakman ook informatie over de toe te dienen dosis. In par. [0075] staat dat een TTS met een geladen dosis rivastigmine van 36 mg waarvan 12 mg wordt afgegeven goed wordt verdragen. Uit par. [0077] van US 938 kent de gemiddelde vakman het orale doseringsregime dat aanvangt bij tweemaal daags 1,5 mg (oftewel 3 mg per dag) en via titratie kan worden opgevoerd tot tweemaal daags 3 mg, 4,5 mg of 6 mg (of te wel daags 6, 9 of 12 mg). Het behoorde tot de algemene vakkennis van de gemiddelde vakman dat een behandeling met rivastigmine gelet op de bijwerkingen altijd een titratieschema kende en dat die titratie aanving met een startdosering. Uit de Exelon SmPC, waarmee de gemiddelde vakman op de prioriteitsdatum volgens Novartis ook bekend was, weet hij dat bij een oraal doseringsregime wordt aangevangen met een dosis van tweemaal daags 1,5 mg die niet therapeutisch effectief is en dat de opvolgende doses wel therapeutisch effectief zijn.
4.27.
Vervolgens leest de vakman in par. [0077] van US 938: “Tolerability is seen to be even better for the transdermal device, wherein 24 mg were absorbed in 24 hours”. De vakman weet op basis van zijn algemene vakkennis dat een korte titratieprocedure en het gebruik van een therapeutisch effectieve aanvangsdosis wenselijk zijn. Novartis heeft onvoldoende gemotiveerd weersproken dat die twee uitgangspunten de therapietrouw bevorderen, omdat ze de toepassing vereenvoudigen en de patiënt direct de gunstige werking van de therapie ervaart. De bijwerkingen van rivastigmine zijn vervelend maar niet zeer gevaarlijk voor patiënten, zodat de vakman bij het testen op zoek zal gaan naar een aanvangsdosering die het optimum biedt tussen therapeutische werking en een acceptabel niveau van bijwerkingen. De hiervoor geciteerde zin in par. [0077] die, in vergelijking met het doseringsschema bij oraal toe te dienen rivastigmine, een hoge, goed verdraagbare dosering na titratie openbaart, zal hem daarom aansporen om bij de voorgestelde routine bioavailability tests te testen of het mogelijk is een hogere aanvangsdosering voor de TTS te gebruiken dan voor orale toediening is vermeld. Gelet op de voordelen die een therapeutische dosis bevat, zal hij daarbij onderzoeken of een therapeutisch effectieve aanvangsdosis ook nog een acceptabel niveau van bijwerkingen kent. De rechtbank verwerpt dan ook de stelling van Novartis dat de vakman - par. [0076] lezende - op zoek zou gaan naar een aanvangsdosering voor een TTS die overeenstemt met de niet-therapeutisch effectieve aanvangsdosis van het orale doseringsregime.
4.28.
Anders dan Novartis (in het verlengde van haar deskundige Fahr ) stelt, zullen de EMA Guidelines de vakman niet weerhouden om bij het ‘routine’ testen de therapeutisch effectieve orale dosering van 6 mg daags te testen. Novartis heeft in dat kader gewezen op het voorschrift in de EMA Guidelines dat “the marketed immediate release product of the same active substance should serve as the reference product”. Alvogen c.s. heeft vervolgens onweersproken aangevoerd dat uit die regel niet volgt dat geen andere doses mogen worden gehanteerd. Dat volgt uit EMA Guidelines regels 281-283 waarin staat dat andere “sterktes” voor de transdermale doseringsvorm toelaatbaar zijn mits zulks (uit een oogpunt van veiligheid ter voorkoming van medicatie-fouten) duidelijk wordt vermeld in de SmPC, de patiëntenbijsluiter en de etikettering van het TTS product. Ook de EMA Guidelines zullen de vakman derhalve niet weerhouden te testen op de therapeutisch effectieve aanvangsdosis die bioequivalent is aan een orale dosis van 6 mg rivastigmine per dag, de laagste therapeutisch effectieve dosis bij oraal toe te dienen rivastigmine. Datzelfde geldt voor de door Novartis – eerst bij pleidooi – aangehaalde passage in de publicatie van Desai (productie EP26v, p. 573) welke passage eindigt (hetgeen Novartis gemakshalve achterwege laat in haar citaat) met de aanbeveling: “It is recommended that patients should receive the highest tolerated dosage.” Gezien die aanbeveling zal de vakman er niet van worden weerhouden te testen wat die highest tolerated dosage bij een aanvangsdosering zou zijn.
4.29.
De rechtbank volgt Alvogen c.s. tot slot in haar stelling dat ook zijn algemene vakkennis de gemiddelde vakman op de prioriteitsdatum niet zou hebben weerhouden, integendeel, om bij de routine tests uit te gaan van de laagste therapeutisch effectieve orale dosis. Onder verwijzing naar de twee verklaringen van haar deskundige Dr. Rue (producties GP08 en GP30) heeft Alvogen c.s. aangevoerd dat op de prioriteitsdatum algemeen werd aangenomen dat de bijwerkingen van rivastigmine samenhangen met de snelle fluctuaties en hoge piekconcentraties (uitgedrukt in Tmax en Cmax) rivastigmine in het bloed na orale toediening. Rue verwijst naar het handboek van Aulton uit 2002 (bijlage 1 bij GP30) waarin dit staat beschreven voor geneesmiddelen in het algemeen. Ook verwijst Rue naar de publicaties van Moriearty et al uit 1993 (bijlage 2 bij GP08) en van Imbimbo uit 2001 (bijlage 6 bij GP08) waaruit volgt dat de bijwerkingen van rivastigmine in relatie staan tot ‘rapid initial peaks of inhibition’ , i.e. het percentage van remming van AChE, welke remming uiteraard groter is bij hogere concentraties rivastigmine, anders gezegd bij een hoge Cmax en een korte Tmax. Daarnaast verwijst Rue naar een overzichtsartikel van Lefèvre uit 2006 (productie EP26e), dus van na de prioriteitsdatum, maar waarin een passage is opgenomen aan het einde waarvan door middel van een voetnoot met nummer 8 wordt verwezen naar een publicatie van Jann, Shirley en Small (bijlage 4 bij GP30)van vóór de prioriteitsdatum:
“The incidence of centrally induced cholinergic gastrointestinal side effects with rivastigmine has been associated with the high maximum plasma concentrations (Cmax) and short times to Cmax (tmax) provided by oral administration. Measures that prolong tmax and reduce Cmax, such as the administration of rivastigmine capsules with food, may improve tolerability of cholinesterase inhibitors 8 9’
In die tijdige publicatie wordt beschreven:
“Another factor is drug absorption rate, since cholinestearase inhibitors that are rapidly absorbed may cause a rapid stimulation of the cholinergic system with adverse effects closely following. Administering the drug with food can lower the amount of gastrointestinal effects by delaying tmax.”
Op grond van het vorenstaande zou de gemiddelde vakman aannemen dat bij transdermale toediening de Cmax zou afnemen zodat een hogere aanvangsdosering dan die bij oraal gebruik niet zonder meer tot problemen zou leiden. In ieder geval bestond er geen vooroordeel die hem daarvan zou hebben weerhouden. Dat bij transdermale toediening het zogenaamde first pass effect niet optreedt en leidt tot een toename van de AUC zou daaraan evenmin in de weg staan, nu, zoals Dr. Rue heeft verklaard, op de prioriteitsdatum algemeen werd aangenomen dat het verhogen van de AUC niet zou leiden tot een afname van de verdraagbaarheid van het geneesmiddel.
4.30.
Het andersluidende betoog van Novartis, onder meer gebaseerd op de verklaring van haar deskundige Fahr en de publicaties van Sramek et al uit 1996 (EP30.2) en Gourlay uit 2001 (EP30.3), dat de vakman een en ander niet zou proberen omdat niet de Cmax maar de AUC verantwoordelijk zou zijn voor bijwerkingen, is gelet op de gemotiveerde betwisting door Alvogen c.s., onvoldoende onderbouwd en wordt verworpen.
4.31.
Al met al zou de gemiddelde vakman zich derhalve realiseren dat het toedienen van een bepaalde dosis rivastigmine middels een TTS door de geleidelijke afgifte aan de patiënt zou leiden tot een lagere Cmax en een verlenging van Tmax ten opzichte van orale toediening van diezelfde dosis met mogelijk minder bijwerkingen als gevolg, maar wel met hetzelfde therapeutisch effect als een vergelijkbare AUC wordt bereikt. De gemiddelde vakman zou dan ook geneigd zijn de routine tests uit te voeren zoals in US 938 vermeld. Van een vooroordeel daartegen was, als gezegd, geen sprake.
4.32.
De gemiddelde vakman zal daarbij een redelijke verwachting van succes hebben omdat in US 938 een voorbeeld staat van (de structuur en samenstelling van) een TTS van 10 cm² met een geladen dosis van 18 mg rivastigmine en hij uit US 938 en de SmPC bekend is met de laagste therapeutisch effectieve orale aanvangsdosering van 6 mg daags op basis waarvan hij kan testen en de tests eenvoudig en routinematig in een kleine klinische studie kunnen worden uitgevoerd. Dat er nog inventieve arbeid schuilt in de keuze voor de maat van de TTS als na testen de aanvangsdosering van de TTS (en daarmee ook de te laden dosering) bekend is, is door Novartis terecht niet aangevoerd.
4.33.
Gelet op het voorgaande zal de gemiddelde vakman naar het oordeel van de rechtbank na kennisneming van US 938 door middel van routinetests op ‘trial and error’ basis komen tot de aanvangsdosering van EP 219 op basis van een TTS van 5 cm² met een geladen dosis van 9 mg rivastigmine. De oplossing voor het objectieve probleem vloeit derhalve op voor de hand liggende wijze voort uit de meest nabije stand van de techniek.
4.34.
De conclusie is dat EP 219 niet inventief en dus niet geldig is. Aangezien op een nietig octrooi geen inbreuk kan worden gemaakt, moeten de vorderingen van Novartis worden afgewezen.
4.35.
Tot slot zij vermeld dat de Engelse rechter in de in 2.13 beschreven parallelle Engelse procedure in vergelijkbare zin oordeelde, zodat die beslissing geen aanleiding geeft tot nadere overweging. De Engelse rechter heeft de inventiviteit van EP 219 beoordeeld aan de hand van US 031 (behorende tot dezelfde octrooifamilie als US 938) waarin een TTS op basis van rivastigmine en een anti-oxidant is beschreven en waarvan niet in geschil is dat de inhoud in feite overeenkomt met die van US 938. Hij komt tot de conclusie (in par. 134 van de uitspraak) dat het voor een vakman voor de hand lag om de dosis afgegeven door een 5 cm² TTS#2 uit te proberen als aanvangsdosis.
Slotsom en proceskosten
4.36.
De overige door Alvogen c.s. aangevoerde nietigheidsgronden en het verweer ten aanzien van de gestelde inbreuk kunnen gelet op het voorgaande onbesproken blijven.
4.37.
Novartis zal als de in het ongelijk gestelde partij worden veroordeeld in de proceskosten aan de zijde van Alvogen c.s. Alvogen c.s. vordert onder verwijzing naar artikel 1019h Rv een vergoeding van haar volledige proceskosten, die zij heeft begroot op € 206.703,09. De redelijkheid en evenredigheid van die kosten heeft Novartis niet bestreden, zodat die kosten zullen worden toegewezen.
5. De beslissing
De rechtbank
5.1.
wijst de vorderingen af;
5.2.
veroordeelt Novartis in de proceskosten aan de zijde van Alvogen c.s. tot op heden begroot op € 206.703,09;
5.3.
verklaart de kostenveroordeling uitvoerbaar bij voorraad;
Dit vonnis is gewezen door mr. M.P.M. Loos, mr. J.Th. van Walderveen en mr. F.M. Bus en in het openbaar uitgesproken op 6 juli 2016.