Hof Den Haag, 10-06-2014, nr. 200.108.409/01
Uitspraak 10‑06‑2014
Mrs. A.D. Kiers-Becking, M.Y Bonneur, C.J.J. van Loon
Partij(en)
arrest van 10 juni 2014
inzake
- 1.
SANDOZ B.V.,
gevestigd te Almere,
- 2.
de vennootschap naar vreemd recht HEXAL AG,
gevestigd te Holzkirchen, Duitsland,
hierna tezamen te noemen (in enkelvoud): Sandoz,
appellanten, incidenteel geïntimeerden,
procesadvocaat: mr. D. Knottenbelt te Rotterdam,
behandelend advocaten: mrs. M.G.R. van Gardingen en N. Wiersma,
tegen
de vennootschap naar vreemd recht ASTRAZENECA AB,
gevestigd te Södertalje, Zweden,
geïntimeerde, incidenteel appellante,
hierna te noemen: AZ,
procesadvocaat: mr. L.Ph.J. van Utenhove te Den Haag,
behandelend advocaat: mr. W.A. Hoyng.
Het geding
Bij exploten van 11 mei 2012 is Sandoz in hoger beroep gekomen van het door de rechtbank 's‑Gravenhage tussen partijen gewezen vonnis van 7 maart 2012. Sandoz heeft bij memorie van grieven negen grieven tegen het vonnis aangevoerd. AZ heeft bij memorie van antwoord, tevens memorie van grieven in (voorwaardelijk) incidenteel appel de grieven bestreden en, (voorwaardelijk) incidenteel appellerende, één grief tegen het vonnis aangevoerd, welke grief door Sandoz bij memorie van antwoord in (voorwaardelijke) incidenteel appel is bestreden. Vervolgens hebben partijen op 16 januari 2014 hun standpunten doen bepleiten door hun voormelde (behandelend) advocaten. Door het hof zijn de volgende, op voorhand gezonden, producties ontvangen:
- —
namens Sandoz een akte met producties 35 tot en met 39 op 2 januari 2014;
- —
namens AZ een akte met producties 44 tot en met 47 op 3 januari 2014.
Nu tegen het in het geding brengen daarvan geen bezwaar is gemaakt, zullen deze worden toegelaten. Voorts heeft het hof een op 15 januari 2014 bij het hof binnengekomen brief van mr. Wiersma ontvangen, waarin is medegedeeld dat partijen zijn overeengekomen dat de redelijke en evenredige kosten van dit hoger beroep € 125.000,-- bedragen.
Tenslotte is arrest gevraagd.
Beoordeling van het hoger beroep
De feiten
1.
De door de rechtbank in rechtsoverwegingen 3.1 tot en met 3.20 en 3.22 tot en met 3.24 van het vonnis als vaststaand aangemerkte feiten zijn niet bestreden, met dien verstande dat Sandoz een aantal van deze feiten niet volledig of niet relevant acht. Daarvan uitgaande gaat het hof uit van de volgende feiten.
1.1.
AZ, een in Zweden gevestigde onderneming behorend tot het AstraZeneca concern dat zich bezig houdt met de ontwikkeling van innovatieve geneesmiddelen, is houdster van het Europees octrooi EP 0 907 364, voor Sustained release pharmaceutical compositions comprising a dibenzothiazepine derivative (hierna: EP 364 of het octrooi). Het octrooi is onder meer van kracht in Nederland.
1.2.
EP 364 is verleend op 14 augustus 2002 op basis van een aanvrage van 27 mei 1997 (hierna: de indieningsdatum) en doet een beroep op prioriteit van de Engelse octrooiaanvrage GB 96 11328 (hierna: GB 328) van 31 mei 1996 (hierna: de prioriteitsdatum). Er is geen oppositie ingesteld tegen de verlening van EP 364.
1.3.
De conclusies van EP 364 luiden in de authentieke (Engelse) taal als volgt.
- ‘1.
A sustained release formulation comprising a gelling agent and 11-[4-[2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo-[b,f][1,4]thiazepine or a pharmaceutically acceptable salt thereof, together with one or more pharmaceutically acceptable excipients.
- 2.
A sustained release formulation according to claim 1 such that 11-[4-[2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo-[b,f][1,4]thiazepine or a pharmaceutically acceptable salt thereof is released from the formulation, in a controlled fashion over a period of between 8 and 24 hours so that at least 60% of 11-[4-[2-(2-hydroxyethoxy) ethyl]-1-piperazinyl]dibenzo-[b,f][1,4]thiazepine or a pharmaceutically acceptable salt thereof has been released at the end of this period.
- 3.
A sustained release formulation according to claim 1 or claim 2 wherein the gelling agent is hydroxypropyl methylcellulose.
- 4.
A sustained release formulation according to claim 3 comprising about 5 to 50% by weight of a hydroxypropyl methylcellulose selected from the group consisting of (a) a hydroxypropyl methylcellulose having a viscosity of about 40 to 60 cps, a methoxy content of about 28 to 30% by weight and a hydroxypropoxy content of from about 7 to less than 9% by weight, (b) a hydroxypropyl methylcellulose having a viscosity of about 3,500 to 5,600 cps, a methoxy content of about 28 to 30% by weight and a hydroxypropoxy content of about 7 to 12% by weight, (c) a hydroxypropyl methylcellulose having a viscosity of about 80 to 120 cps, a methoxy content of about 19 to 24% by weight and a hydroxypropoxy content of from about 7 to less than 9% by weight and (d) a hydroxypropyl methylcellulose having a viscosity of about 3,500 to 5,600 cps, a methoxy content of about 19 to 24% by weight and a hydroxypropoxy content of about 7 to 12% by weight, or mixtures thereof.
- 5.
A sustained release formulation according to claim 3 comprising about 5 to 50% by weight of a hydroxypropyl methylcellulose selected from the group consisting of (a) a hydroxypropyl methylcellulose having a viscosity of about 40 to 60 cps, a methoxy content of about 28 to 30% by weight and a hydroxypropoxy content of from about 7 to less than 9% by weight, (b) a hydroxypropyl methylcellulose having a viscosity of about 3,500 to 5,600 cps, a methoxy content of about 28 to 30% by weight and a hydroxypropoxy content of about 7 to 12% by weight, (c) a hydroxypropyl methylcellulose having a viscosity of about 80 to 120 cps, a methoxy content of about 19 to 24% by weight and a hydroxypropoxy content of from about 7 to less than 9% by weight and (d) a hydroxypropyl methylcellulose having a viscosity of about 3,500 to 5,600 cps, a methoxy content of about 19 to 24% by weight and a hydroxypropoxy content of about 7 to 12% by weight, or mixtures thereof with the proviso that if the formulation contains a hydroxypropyl methylcellulose described under (d) above the total amount of hydroxypropyl methylcellulose present in the formulation must be greater than 25.8% by weight.
- 6.
A sustained release formulation according to claim 4 or claim 5 comprising about 5 to 40% by weight of a hydroxypropyl methylcellulose selected from the group consisting of (a) – (d) or mixtures thereof.
- 7.
A sustained release formulation according to claim 6 comprising about 8 to 35% by weight of a hydroxypropyl methylcellulose selected from the group consisting of (a) – (d) or mixtures thereof.
- 8.
A formulation according to claim 7 comprising about 10 to 30% by weight of a hydroxypropyl methylcellulose selected from the groups (a) – (d) or mixtures thereof.
- 9.
A formulation according to claim 8 comprising about 15 to 30% by weight of a hydroxypropyl methylcellulose selected from the groups (a) – (d) or mixtures thereof.
- 10.
A formulation according to anyone of claims 1–9 wherein 11-[4-[2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo[b,f][1,4]thiazepine or a pharmaceutically acceptable salt thereof is present in about 35 to 65% by weight.
- 11.
A formulation according to claim 10 wherein the amount of hydroxypropyl methylcellulose is about 5 to 40%.
- 12.
A formulation according to claims 1–11 wherein the one or more pharmaceutically acceptable excipients are selected from the group consisting of microcrystalline cellulose, lactose, magnesium stearate, sodium citrate and povidone.
- 13.
A formulation according to claim 12 wherein the one or more pharmaceutically acceptable excipients are selected from the group consisting of (a) about 4 to 20% by weight of microcrystalline cellulose, (b) about 5 to 20% by weight of lactose, (c) about 1 to 3% by weight of magnesium stearate, (d) about 10 to 30% by weight of sodium citrate 15 and (e) about 1 to 15% by weight of povidone.
- 14.
A formulation according to anyone of claims 1–13 wherein one of the one or more pharmaceutically acceptable excipients is a pH modifier.
- 15.
A formulation according to claim 14 wherein the pH modifier is sodium citrate.
- 16.
A formulation according to any of claims 1–15 wherein 11-[4-[2-(2-hydroxyethoxy) ethyl]-1-piperazinyl]dibenzo[b,f] [1,4]thiazepine is in the form of a hemifumarate salt.
- 17.
A formulation according to any one of claims 1–16 wherein the formulation is coated.
- 18.
The use of a formulation according to any one of claims 1–17 in the manufacture of a medicament for treating psychotic states or hyperactivity in a warm-blooded animal.
- 19.
A process for preparing a formulation according to any one of claims 1–17 which comprises mixing 11-[4-[2-(2-hydroxyethoxy) ethyl]-1-piperazinyl] — dibenzo[b,f][11,4]thiazepine, or a pharmaceutically acceptable salt thereof, a gelling agent and other excipients.
- 20.
A process for preparing a formulation according to any one of claims 1–17 which comprises:
- a)
mixing 11-[4-[2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]-dibenzo[b,f][1,4]thiazepine, or a pharmaceutically acceptable salt thereof, a gelling agent and other excipients;
- b)
wet granulating the mixed components;
- c)
drying the mixture;
- d)
milling the dried mixture;
- e)
blending the mixture with a lubricant; and
- f)
compressing the blended mixture to form tablets, and optionally coating said tablets.’
1.4.
De conclusies van EP 364 luiden in de Nederlandse vertaling als volgt.
- ‘1.
Formulering voor gereguleerde afgifte omvattende een geleermiddel en 11-[4-][2-(2-hydroxyethoxy)ethyl]-1-piperazinyl] dibenzo[b,f] [1,4]-thiazepine of een farmaceutisch aanvaardbaar zout daarvan, samen met één of meer farmaceutisch aanvaardbare excipiënten.
- 2.
Formulering voor gereguleerde afgifte volgens conclusie 1 zodanig dat 11-[4-][2-(2-hydroxyethoxy)ethyl]-1 piperazynyl] dibenzo[b,f][1,4]-thiazepine of een farmaceutisch aanvaardbaar zout daarvan uit de formulering wordt afgegeven op een geregelde wijze gedurende een periode tussen 8 en 24 uur zodanig dat ten minste 60% van 11-[4-][2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo[b,f][1,4]-thiazepine of een farmaceutisch aanvaardbaar zout daarvan aan het einde van deze periode is afgegeven.
- 3.
Formulering voor gereguleerde afgifte volgens conclusie 1 of conclusie 2, waarbij het geleermiddel hydroxypropylmethylcellulose is.
- 4.
Formulering voor gereguleerde afgifte volgens conclusie 3, omvattende ongeveer 5 tot 50% gew.% van een hydroxypropylmethylcellulose gekozen uit de groep bestaande uit (a) een hydroxypropylmethylcellulose met een viscositeit van ongeveer 40 to 60 cps, een methoxygehalte van ongeveer 28 to 30% gew.% en een hydroxypropoxygehalte van ongeveer 7 tot kleiner dan 9 gew.%; (b) een hydroxypropylmethylcellulose met een viscositeit van ongeveer 3500 tot 5600 cps, een methoxygehalte van ongeveer 28 tot 30 gew.% en een hydroxypropoxygehalte van ongeveer 7 tot 12 gew.%; (c) een hydroxypropylmethylcellulose met een viscositeit van ongeveer 80 tot 120 cps, een methoxygehalte van ongeveer 19 tot 24 gew.% en een hydroxypropoxy-gehalte van ongeveer 7 tot kleiner dan 9 gew.% en (d) een hydroxypropylmethylcellulose met een viscositeit van ongeveer 2500 tot 5600 cps, een methoxygehalte van ongeveer 19 tot 24 gew.% en een hydroxypropoxygehalte van ongeveer 7 tot 12 gew.%, of mengsels daarvan.
- 5.
Formulering voor gereguleerde afgifte volgens conclusie 3, omvattende ongeveer 5 tot 50 gew.% van een hydroxypropylmethylcellulose gekozen uit de groep bestaande uit (a) een hydroxypropylmethylcellulose met een viscositeit van ongeveer 40 tot 60 cps, een methoxygehalte van ongeveer 28 tot 30 gew.% en een hydroxypropoxygehalte van ongeveer 7 tot kleiner dan 9 gew.%; (b) een hydroxypropylmethylcellulose met een viscositeit van ongeveer 3500 tot 5600 cps, een methoxygehalte van ongeveer 28 tot 30 gew.% en een hydroxypropoxygehalte van ongeveer 7 tot 12 gew.%; (c) een hydroxypropylmethylcellulose met een viscositeit van ongeveer 80 tot 120 cps, een methoxygehalte van ongeveer 19 tot 24 gew.% en een hydroxypropoxygehalte van ongeveer 7 tot kleiner dan 9 gew.% en (d) een hydroxypropylmethylcellulose met een viscositeit van ongeveer 3500 tot 5600 cps, een methoxygehalte van ongeveer 19 tot 24 gew.% en een hydroxypropoxygehalte van ongeveer 7 tot 12 gew.%, of een mengsels daarvan met dien verstande dat als de formulering een hydroxypropylmethylcellulose beschreven onder (d) hierboven bevat de totale hoeveelheid hydroxypropylmethylcellulose aanwezig in de formulering groter dan 25,8 gew.% moet zijn.
- 6.
Formulering voor gereguleerde afgifte volgens conclusie 4 of conclusie 5, omvattende ongeveer 5 tot 40 gew.% van een hydroxypropylmethylcellulose gekozen uit de groep bestaande uit (a) – (d) of mengsels daarvan.
- 7.
Formulering voor gereguleerde afgifte volgens conclusie 6, omvattende ongeveer 8 tot 35 gew.% van een hydroxypropylmethylcellulose gekozen uit de groep bestaande uit (a) – (d) of mengsels daarvan.
- 8.
Formulering voor gereguleerde afgifte volgens conclusie 7, omvattende ongeveer 10 tot 30 gew.% van een hydroxypropylmethylcellulose gekozen uit de groep bestaande uit (a) – (d) of mengsels daarvan.
- 9.
Formulering voor gereguleerde afgifte volgens conclusie 8, omvattende ongeveer 15 tot 30 gew.% van een hydroxypropylmethylcellulose gekozen uit de groep bestaande uit (a) – (d) of mengsels daarvan.
- 10.
Formulering volgens één der conclusies 1–9, waarbij 11-[4-][2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo[b,f][1,4]-thiazepine of een farmaceutisch aanvaardbaar zout daarvan aanwezig is in ongeveer 35 tot 65 gew.%.
- 11.
Formulering volgens conclusie 10, waarbij de hoeveelheid hydroxypropylmethylcellulose ongeveer 5 tot 40% is.
- 12.
Formulering volgens één der conclusies 1–11, waarbij de één of meer farmaceutisch aanvaardbare excipiënten worden gekozen uit de groep bestaande uit microkristallijne cellulose, lactose, magnesiumstearaat, natriumcitraat en Povidone.
- 13.
Formulering volgens conclusie 12, waarbij de één of meer farmaceutisch aanvaardbare excipiënten worden gekozen uit de groep bestaande uit (a) ongeveer 4 tot 20 gew.% microkristallijne cellulose, (b) ongeveer 5 tot 20 gew.% lactose, (c) ongeveer 1 tot 3 gew.% magnesiumstearaat, (d) ongeveer 10 tot 30 gew.% natriumcitraat en (e) ongeveer 1 tot 15 gew.% Povidone.
- 14.
Formulering volgens één der conclusies 1–13, waarbij de één of meer farmaceutisch aanvaardbare excipiënten een pH-modificator is.
- 15.
Formulering volgens conclusie 14, waarbij de pH-modificator natriumcitraat is.
- 16.
Formulering volgens één der conclusies 1–15, waarbij 11-[4-][2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo[b,f][1,4]-thiazepine in de vorm van een hemifumaraatzout is.
- 17.
Formulering volgens één der conclusies 1–16, waarbij de formulering bekleed is.
- 18.
Toepassing van een formulering volgens één der conclusies 1–17 bij de productie van een geneesmiddel voor het behandelen van psychotische toestanden of hyperactiviteit bij een warmbloeding dier.
- 19.
Werkwijze voor het bereiden van een formulering volgens één der conclusies 1–17, welke omvat het mengen van 11-[4-][2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo[b,f][1,4]-thiazepine, of een farmaceutisch aanvaardbaar zout daarvan, een geleermiddel en andere excipiënten.
- 20.
Werkwijze voor het bereiden van een formulering volgens één der conclusies 1–17, welke omvat:
- a)
het mengen van 11-[4-][2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo[b,f][1,4]-thiazepine, of een farmaceutisch aanvaardbaar zout daarvan, een geleermiddel, en andere excipiënten;
- b)
het nat granuleren van de gemengde bestanddelen;
- c)
het drogen van het mengsel;
- d)
het malen van het gedroogde mengsel;
- e)
het mengen van het mengsel met een smeermiddel; en
- f)
het comprimeren van het gemengde mengsel om tabletten te vormen, en eventueel het bekleden van genoemde tabletten.’
1.5
De stof 11-[4-][2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo[b,f][1,4]-thiazepine is bekend onder de naam quetiapine.
1.6
EP 240 228 (hierna: EP 228), ook van AZ, is het basisoctrooi voor de stof quetiapine en is per 24 maart 2007 verlopen. Op basis van dit octrooi is een aanvullend beschermingscertificaat verleend (980022) dat op 23 maart 2012 is verlopen. Dit basisoctrooi is gepubliceerd in 1987.
In de octrooibeschrijving van EP 364 is vermeld dat quetiapine ‘may be used , for example, as an antipsychotic agent (for example for the management of the manifestations of psychotic disorders) or as a treatment for hyperactivity’ (par. 0007). Quetiapine is een zogenaamd atypisch antipsychoticum.
1.7
In de (door Sandoz overgelegd als productie — hierna: S — 12 en door AZ als productie — hierna: A -21A) publicatie van O. Gefvert e.a, ‘Time course for dopamine and serotonin receptor occupancy in the brain of schizophrenic patients following dosing with 150 mg Seroquel™ tid.’, abstract P-4-65, gepresenteerd op the 8th European College of Neuropsychopharmacology Congress in Venice in 1995 (hierna: Gefvert), is onder meer het volgende vermeld:
‘SEROQUEL™ (…) is an atypical dibenzothiazepine antipsychotic agent in Phase III development by Zeneca Pharmaceuticals. Dosing of SEROQUEL in the Phase II/III programme was TID [driemaal daags — hof] and QID [viermaal daags — hof], based partly on preliminary pharmacokinetic data for the parent compound (Tmax approximately 1,5 h, plasma elimination half-life approximately 3 h). Given the importance of compliance with medication in schizophrenics, a more convenient dose regimen would be beneficial.
This was an open, non-randomised trial to determine whether receptor occupancy data is consistent with BID [tweemaal daags — hof] dosing. Schizophrenic patients (DSM IIIR) were dosed with 150 mg SEROQUEL three times a day (…). There were no serious adverse events. No extrapyramidal side effects (EPS) were reported during the dosing phase. (…) Mean plasma elimination half-life was approximately 5.3 hours (range 2.7–9.3 hours).
Once to twice daily dosing may therefore maintain sufficient 5HT2/D2 receptor occupancy for therapeutic benefit with a low incidence of EPS in schizophrenic patients. A large efficacy study in 622 patients (SAFARI) comparing BID and TID regimens is in progress.’
1.8
De in EP 364 geoctrooieerde uitvinding ziet, kort gezegd, op een formulering voor gereguleerde afgifte (hierna ook: sustained release of SR) van een samenstelling omvattende een geleermiddel en de werkzame stof quetiapine of een farmaceutisch aanvaardbaar zout daarvan, samen met één of meer farmaceutisch aanvaardbare excipiënten, alsmede de toepassing van een dergelijke formulering bij de productie van een geneesmiddel en een werkwijze voor de bereiding ervan.
1.8
De beschrijving van EP 364 bevat onder meer de navolgende passages:
‘(…)
- [0002]
It is desirable in the treatment of a number of diseases, both therapeutically and prophylactically, to provide an active pharmaceutical ingredient in a sustained release form. Desirably the sustained release provides a generally uniform and constant rate of release over an extended period of time which achieves a stable and desired blood (plasma) level of the active ingredient without the need for frequent administration of the medicament.
- [0003]
While there are numerous sustained release formulations known in the art which utilize gelling agents, such as hydroxypropyl methylcelluloses, it has been found to be difficult to formulate sustained release formulations of soluble medicaments and gelling agents, such as hydroxypropyl methylcellulose, for several reasons. First of all, active ingredients which are soluble in water tend to generate a sustained release product which is susceptible to a phenomenon known as dose dumping. That is, release of the active ingredient is delayed for a time but once release begins to occur the rate of release is very high. Moreover, fluctuations tend to occur in the plasma concentrations of the active ingredient which increases the likelihood of toxicity. Further, some degree of diurnal variation in plasma concentration of the active ingredient has also been observed. Finally, it has been found to be difficult to achieve the desired dissolution profiles or to control the rate of release of the soluble medicament.
- [0004]
Accordingly, a need exists for sustained release formulations of soluble medicaments, such as 11-[4-][2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]-dibenzo[b,f][1,4]thiazepine or a pharmaceutically acceptable salt, which overcome, or at least alleviate, one or more of the above described difficulties and which further provide the advantageous property of allowing the active medicament to be administered less frequently, e.g. once a day, while achieving blood (plasma) levels similar to those attained by administering smaller doses of the medicament more frequently, e.g. two or more times daily. (…)
- [0007]
The compound (…) and its pharmaceutically acceptable salts exhibit useful antidopaminergic activity and may be used, for example, as an antipsychotic agent (for example, for the management of the manifestations of psychotic disorders) or as a treatment for hyperactivity. (…)
- [0008]
The preparation, physical properties and beneficial pharmacological properties of 11-[4-][2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]-dibenzo[b,f][1,4]thiazepine, and its pharmaceutically acceptable salts are described in published European patents EP 240,228 and 282,236 as well as in U.S. Patent 4,879,288.’
1.9
AZ brengt een farmaceutisch product met het actief ingrediënt quetiapine op de markt onder de merknaam Seroquel, thans zowel in een onmiddellijke afgifte (IR) formulering (Seroquel IR) als in een vertraagde afgifte SR formulering (Seroquel XR). Zowel Seroquel IR als Seroquel XR is toegelaten voor de behandeling van schizofrenie en bipolaire stoornis. Seroquel XR is daarnaast ook geïndiceerd voor de behandeling van Major Depressive Disorders (MDD).
De vorderingen
2.
In conventie heeft Sandoz vernietiging van het Nederlands deel van EP 364 gevorderd. Sandoz voert daartoe aan dat alle conclusies van EP 364 inventiviteit ontberen. In hoger beroep zijn nog slechts de conventionele vorderingen aan de orde, nu de reconventionele vorderingen van AZ zijn afgewezen en daartegen geen (incidenteel) beroep is ingesteld.
3.
AZ heeft gemotiveerd verweer gevoerd, primair stellende dat het octrooi geldig is, subsidiair dat het geldig is conform hulpverzoek 1 en meer subsidiair dat het geldig is op basis van hulpverzoek 2.
De hulpverzoeken (A 20A en 20B) van luiden als volgt:
Hulpverzoek 1:
‘A sustained release formulation in a tablet form, comprising a gelling agent and an effective dosage amount of 11-[4-[2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo-[b,f][1,4]thiazepine or a pharmaceutically acceptable salt thereof, together with one or more pharmaceutically acceptable excipients wherein
- —
11-[4-[2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo-[b,f][1,4]thiazepine or a pharmaceutically acceptable salt thereof is released from the formulation, in a controlled fashion over a period of 8 hours or longer, and wherein
- —
11-[4-[2-(2-hydroxyethoxy)ethyl]-1-piperazinyl]dibenzo[b,f][1,4]thiazepine or a pharmaceutically acceptable salt thereof is present in about 35 to 65% by weight.’
Hulpverzoek 2 voegt daaraan verder toe:
- ‘—
and wherein the plasma concentration of the active ingredient over time profile (AUC) for the sustained release formulation is about the same as for an immediate release formulation.’
4.
De rechtbank heeft het in conventie gevorderde afgewezen.
5.
De principale grieven 1 tot en met 7 richten zich tegen het oordeel dat het octrooi inventief is en de daarvoor gegeven motivering. Grief 8 richt zich tegen de feitenvaststelling door de rechtbank. Grief 9 is een restgrief, die zich richt tegen de afwijzing van de conventionele vorderingen en de veroordeling van Sandoz in de kosten van de procedure. De incidentele grief richt zich tegen de verwerping door de rechtbank in rechtsoverweging 5.46 van een door AZ aangevoerd verweer tegen de conventionele vorderingen.
De algemene vakkennis
6.
Het hof gaat, naast de niet bestreden feitenvaststelling door de rechtbank, uit van de volgende, enerzijds gestelde en anderzijds erkende of niet gemotiveerd betwiste, algemene vakkennis van de gemiddelde vakman op de prioriteitsdatum. Daarbij heeft het onder meer gebruik gemaakt van de (formulerings)handboeken van Remington, The Science and Practice of Pharmacy, hoofdstuk 94: sustained-Release Drug Delivery Systems (editie 1995) (S 6) — hierna: Remington — , M.E. Aulton, Pharmaceutics: The Science of Dosage Form Design 1988, hoofdstuk 18: Tablets (S 7) — hierna: Aulton —, L. Lachman e.a., The Theory and Practice of Industrial Pharmacy, Hoofdstuk 14: Sustained Release Dosage Forms (editie 1987) (S 8) — hierna: Lachman — en de beslissing van the High Court of Justice, Chancery Division Patents Court (Justice Arnold) — hierna: the High Court — over dit zelfde octrooi van 22 maart 2012 (S 22).
Op het gebied van medicatie tegen schizofrenie
7.
Schizofrenie wordt in de regel behandeld met antipsychotica. Antipsychotica blokkeren bepaalde dopaminereceptoren in de hersenen. Dopamine is een neurotransmitter die in verschillende delen van de hersenen wordt aangemaakt en daar vijf bekende dopaminereceptoren activeert (D1–D5). Schizofreniepatiënten hebben een hoger niveau aan dopamine D2-receptoren en subcorticale dopamineactiviteit. Alle klinisch werkzame antipsychotica hebben in meer of mindere mate een blokkerende werking op D2-receptoren.
8.
De eerste antipsychotica, waaronder chloorpromazine, zijn aan het begin van de jaren vijftig ontwikkeld. Het ging daarbij om zogenaamde typische antipsychotica. Deze hadden als ernstige bijwerking extrapiramidale symptomen (EPS). In het begin van de jaren zestig is clozapine, een nieuw antipsychoticum dat geen EPS opwekt, ontwikkeld. Het ging hier om een zogenaamd atypisch antipsychoticum. Dit middel werd echter van de markt gehaald omdat zich in 1% van de gevallen bij gebruik van dit middel een zeer ernstige bijwerking voordeed, namelijk agranulocytose (een acute sterke afname van het aantal witte bloedcellen). Nadat clozapine van de markt was gehaald is het in 1989 of 1990 weer op de markt gebracht (waarbij het bloed van de patienten nauwkeurig in de gaten moet worden gehouden). Voorts zijn toen andere antipsychotica ontwikkeld, zoals risperidon, olanzapine en quetiapine. Deze zijn in de jaren negentig op de markt zijn gekomen en veroorzaken geen of aanzienlijk minder EPS. Ook hier gaat het om atypische antipsychotica. Op de prioriteitsdatum was risperidon op de markt, terwijl olanzapine (van Lilly) en quetiapine zich in de testfase bevonden.
Op de prioriteitsdatum werd ervan uitgegaan dat voor typische antipsychotica een D2-receptorbezetting van meer dan 60% nodig was om effectief te zijn. Ook was bekend dat dit niet gold voor clozapine en dat voor clozapine een D2-receptorbezetting in de marge van 20 tot 60% voldoende was voor effectiviteit.
9.
Op de prioriteitsdatum was bekend over quetiapine:
- —
er is sprake van een hoog, althans ‘actief first pass metabolisme’ (uit dierenstudies; H. Wetzel e. a., Seroquel (ICI 204 636) a putative ‘atypical’ antipsychotic, in schizophrenia with positive symptomatology: results of an open clinical trial and changes of neuroendocrinological aan EEG parameters, 1995 (A25) — hierna: Wetzel —);
- —
er is sprake van een lage biologische beschikbaarheid (uit dierenstudies; Wetzel); — de halfwaardetijd ligt tussen ongeveer 3 (L.F. Fabre e.a., ICI 204,363, a Novel, Atypical Antipsychotic: Early Indication of Safety and Efficacy in patients with Chronic and Subchronic Schizophrenia, 1995 (A26) — hierna: Fabre —) en 5,3 uur (Gefvert);
- —
de oplosbaarheid varieert van 18,5 mg/ml bij pH 1 tot 2,3 mg/ml bij pH 7.
10.
Vanwege de aard van psychische stoornissen, zoals schizofrenie hebben patiënten veelal moeite zich aan het behandelschema te houden.
Wat betreft SR en IR -formuleringen
11.
Vertraagde afgifte (SR) formuleringen waren op de prioriteitsdatum al bekend, evenals de voordelen die daaraan verbonden zijn. Bij onmiddellijke afgifte (IR) formuleringen wordt de werkzame stof ineens (meest in de maag) vrijgegeven. Hierdoor ontstaat relatief snel een hoge piek van de werkzame stof in het bloed (Cmax) die ook relatief snel weer afneemt. Voordat de werkzame stof in de bloedsomloop komt gaat het door de lever, waar metabolisatie plaats vindt, het zogenaamde ‘first pass metabolism’. Een deel van de werkzame stof bereikt de bloedstroom dus niet. Vervolgens neemt de concentratie in het bloed af bij volgende metabolisaties. De zogenaamde halfwaardetijd is de tijd waarin de helft van de werkzame stof is gemetaboliseerd. De absorptiesnelheid en metabolisatie in de lever zijn bij een IR- formulering bepalend voor de bloedplasmaconcentratie en het verloop daarvan (de plasmacurve). Bij een SR-formulering wordt de werkzame stof langzamer en constanter vrijgegeven als gevolg waarvan doorgaans een stabieler en langduriger plasmaprofiel wordt gerealiseerd. Er is normaliter een minder snelle toename van de plasmaconcentratie (een minder hoge piek) in vergelijking met de onmiddellijke afgifte formulering en de plasmaconcentratie houdt langer aan in tijd (de curve wordt langgerekter). Daarbij is de afgiftesnelheid uit de formulering (mede) bepalend voor de plasmacurve.
12.
Om werkzaam te zijn en geen onacceptabele bijwerkingen te hebben dient de plasmaconcentratie van de werkzame stof in het bloed zich te bevinden tussen een bepaalde onder- en bovengrens. De hieronder afgebeelde tekening uit voormelde uitspraak van de High Court of Justice toont een enkele SR dosis (de rode stippellijn) en drie IR doses. De onderste stippellijn geeft de minimale effectieve concentratie aan (MEC). De bovenste lijn geeft de maximaal veilige concentratie (MSC) aan.
Het doel van iedere formulering is de plasmaconcentratie zo lang mogelijk te houden binnen het therapeutische bereik (de plasmaconcentratiebandbreedte waarbinnen therapeutisch effect maar geen (onaanvaardbare) toxiciteit optreedt).
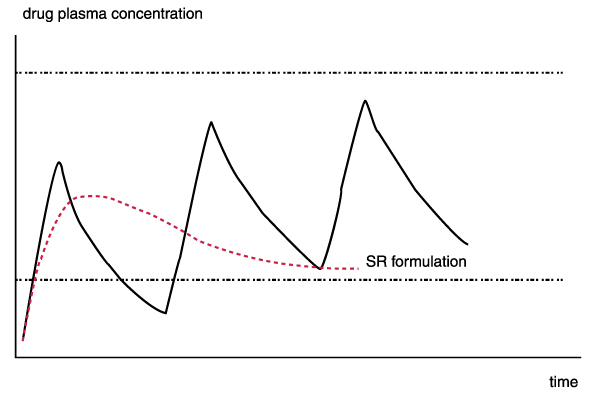
Handhaving van de gewenste plasmaconcentratie binnen het therapeutisch bereik valt onder meer te bereiken door:
- 1.
het verhogen van de dosis, maar dat kan leiden tot toxische waarden;
- 2.
het meerdere malen toedienen van kleinere doses, maar dan is veelal (afhankelijk van de plasma-halfwaardetijd) frequente(re) toediening vereist;
- 3.
het gebruik van een SR-formulering, waarvan een voordeel is dat het de mogelijkheid biedt van een eendaagse, althans minder frequente toediening.
Wanneer de plasma halfwaardetijd lang (meer dan 8 uur) is, bestaat de kans dat de therapeutische plasmaconcentraties gehandhaafd kunnen blijven bij een eenmaal daagse dosis in een IR-formulering. Als de plasma halfwaardetijd korter is zal in het algemeen een SR-formulering nodig zijn om met een eenmalige toediening te kunnen volstaan. SR-formuleringen zijn minder geschikt voor geneesmiddelen met een plasma halfwaardetijd van minder dan 1 of 2 uur of meer dan 8 of 12 uur. SR- formuleringen zijn minder geschikt voor medicijnen met een hoge oplosbaarheid.
13.
Een veel gebruikte methode om vertraagde afgifte te bereiken is — en was op de prioriteitsdatum — diffusie door middel van een zogenaamde reservoir- of matrixformulering. Het was algemeen bekend dat matrixformuleringen betrekkelijk makkelijk zijn te vervaardigen. Als matrixmateriaal kan een hydrofiele polymeer worden gebruik, die een gel vormt. Remington noemt uitdrukkelijk HPMC als geschikt geleermiddel. Hydrofiele matrixmaterialen, waaronder HPMC, waren op de prioriteitsdatum in de handel verkrijgbaar en werden ook gebruikt in SR-formuleringen, (vergelijk de productdocumentatie van Dow Chemical Company U.S.A. over METHOCEL uit 1995, Formulating for Controlled Release with METHOCEL premium cellulose ethers (S 9) — hierna: Dow —). Hydrofiele matrices hebben als voordeel dat 100% van de werkzame stof in vivo wordt afgegeven, hetgeen niet het geval is bij de andere typen matrices. De vertraagde afgifte wordt bereikt door vorming van een gellaag als gevolg van hydratatie van het polymeer. In het lichaam van de patiënt vindt erosie van de (buitenste) gellaag plaats. Op de prioriteitsdatum waren er ook wateroplosbare werkzame stoffen, geformuleerd in een SR-formulering met een hydrofiele matrix, bekend. Vergelijk N.A. Peppas, Hydrogels in Medicine and Pharmacy 1987 (S 11, tabel 14).
Inventiviteit
14.
Inventiviteit ontbreekt als de gemiddelde vakman, uitgaande van de meest nabij stand van de techniek, het probleem zou (would) — en niet slechts kon (could) — hebben opgelost op de wijze geclaimd in het octrooi. Een uitvinding ligt niet alleen voor de hand als de resultaten duidelijk voorspelbaar zijn, maar ook als er sprake is van een redelijke verwachting van succes (reasonable expectation of success), dat wil zeggen dat de vakman in staat is om redelijkerwijs een succesvol einde van een onderzoeksproject binnen acceptabele tijd te voorspellen (Case Law 2013, p. 184 en 185). Onvoldoende is de enkele ‘hope to succeed’. De vakman heeft toegang tot alles in ‘the state of the art’ en heeft ‘the normal means and capacity for routine work and experimentation’ tot zijn beschikking. Hij heeft een conservatieve houding, neemt geen onberekenbare risico's en doet alleen routinematige experimenten (Case Law p. 180–182). Van belang kan zijn of de te verrichten onderzoeken tijdrovend en/of ingewikkeld zijn dan wel er slechts routinematige testen nodig zijn. (Case Law 2013, p. 185).
De gemiddelde vakman
15.
Geen grief is gericht tegen het oordeel van de rechtbank dat de gemiddelde vakman een team is bestaande uit een clinicus op het gebied van psychische stoornissen (psychiater) en een formuleringsdeskundige, zodat het hof daarvan uitgaat.
De relevante datum
16.
Partijen verschillen van mening over de relevante datum. Volgens Sandoz moet worden uitgegaan van de indieningsdatum, 27 mei 1997. Volgens AZ dient te worden uitgegaan van de prioriteitsdatum, 31 mei 1996. Daar dit voor de onderhavige beslissing geen verschil maakt zal het hof hierna uitgaan van de prioriteitsdatum, 31 mei 1996.
De dichtstbijzijnde stand van de techniek
17.
Bij de beoordeling van de inventiviteitsvraag dient de dichtstbijzijnde stand van de techniek of de ‘closest prior art’ (hierna: CPA) te worden vastgesteld op 31 mei 1996. Het hof is met Sandoz van oordeel dat één van de publicaties over quetiapine (IR) als CPA moet worden aangemerkt. Daarbij liggen het EP 228 (waarvan de aanvraag is gepubliceerd in 1987) en Gefvert (1995) voor de hand. Het octrooi noemt EP 228. Gefvert heeft over quetiapine (IR) voor de prioriteitsdatum gepubliceerd. Zowel EP 228 als Gefvert is (ieder op zich) een document ‘disclosing subject matter conceived for the same purpose’ als de uitvinding en ‘having the most relevant technical features in common, i.e. requiring the minimum of structural modifications’ (vergelijk Case Law 2013, p. 167 ev). Dat laatste geldt niet voor de op de prioriteitsdatum gebruikelijke geneesmiddelen tegen schizofrenie, die volgens AZ als CPA moet worden aangemerkt. Het hof kiest voor Gefvert als CPA, omdat daarin ook de daadwerkelijke klinische toepassing van quetiapine (IR) wordt geïllustreerd. Hieruit blijkt niet alleen de stof quetiapine (IR), maar ook de farmacologische eigenschappen daarvan als zijnde een atypisch antipsychoticum.
Problem (and) Solution Approach.
18.
Het hof zal in dit geval als hulpmiddel voor het vaststellen van de inventiviteit de Problem (and) Solution Approach (hierna: PSA) toepassen. AZ stelt dat de PSA in het onderhavige geval niet (noodzakelijk) gebruikt dient te worden, maar geeft niet aan waarom niet en/of dat toepassing tot een onjuiste uitkomst zou leiden. Integendeel zij meent dat met een juiste toepassing van de PSA de, in haar ogen, juiste conclusie getrokken zal worden.
19.
Partijen verschillen over de wijze waarop de PSA in casu moet worden toegepast. Uitgaande van de PSA dient het objectief technische probleem te worden geformuleerd door het verschil tussen de CPA, in casu, kort gezegd, quetiapine (IR) en, kort gezegd, de kenmerkende maatregel(en) in de conclusie(s) van het octrooi vast te stellen en vervolgens daarvan het technisch effect te bepalen. De formulering van het probleem dient enerzijds zo specifiek mogelijk te zijn, maar mag anderzijds geen elementen van of pointers naar de oplossing bevatten (zie Case Law p. 176, T 1019/99 en T 1557/07). Sandoz stelt dat het bedoelde verschil is dat in de conclusie sprake is van een (oraal toe te dienen) formulering voor gereguleerde afgifte (omvattende een geleermiddel, derhalve) door middel van een geleermiddel van quetiapine. Dit wordt door AZ op zichzelf niet betwist, maar zij stelt dat het gaat om een vertraagde afgifteproduct met een PH afhankelijke afgiftesnelheid en een voldoende biologische beschikbaarheid. Dit is echter niet vermeld in de conclusies van het octrooi en hier, bij de vaststelling van de objectieve probleemstelling, niet relevant — later zal dit wel aan de orde komen, zie rechtsoverwegingen 30 ev. In het octrooi is als technisch effect hiervan beschreven dat quetiapine vertraagd wordt afgegeven, hetgeen resulteert in een stabiel en gewenst plasmaniveau (zoals bij het vaker toedienen van een IR-formulering) zonder de noodzaak van frequente toediening van het geneesmiddel. Sandoz heeft het technisch effect omschreven als ‘een stabieler plasmaniveau van quetiapine zonder verhoging van de doseringsfrequentie’. Daarbij gaat zij kennelijk uit van een eenmalige, althans zodanig lage doseringsfrequentie van de IR-formulering, dat daarbij geen stabiel plasmaniveau optreedt. Ook een IR-formulering kan resulteren in een stabiel plasmaniveau, maar dan moet het in beginsel frequenter worden toegediend (zie hiervoor ro. 12). AZ bestrijdt deze omschrijving van het technisch effect en wijst erop dat in paragraaf 0004 van het octrooi sprake is van een formulering met het voordeel ‘allowing the active medicament to be administered less frequently, e.g. once a day’. Gelet daarop is het hof van oordeel dat het technisch effect omschreven moet worden als het bereiken van een stabiel en gewenst plasmaniveau met een minder frequente toediening. Dat zijn immers de in het octrooi genoemde effecten van de SR-formulering. Het octrooi beschrijft geen andere onderscheidende effecten. Het noemt ook geen klinisch effect, anders dan onder verwijzing naar het basisoctrooi, (de stand van de techniek), te weten het gebruik van quetiapine (IR) voor de behandeling van schizofrenie en hyperactiviteit.
AZ stelt dat bij de formulering van het objectieve probleem ook andere technische effecten dan wel onverwachte, niet in het octrooi genoemde, voordelen van het geneesmiddel dienen te worden betrokken. Zij noemt in dit verband dat de uitvinding als voordelen heeft dat het minder bijwerkingen heeft, dat het ook geschikt is voor de behandeling van bipolaire depressie en MDD en dat patiënten veel sneller kunnen worden getitreerd (men kan beginnen met een hogere dosis zonder te erge bijwerkingen, waardoor de gewenste dosis al binnen enkele dagen kan worden gegeven, terwijl dat bij de IR-formulering ten minste zes dagen duurt). Het hof is van oordeel dat het hier — voor zover er al sprake is van daadwerkelijke effecten/voordelen — gaat om bonuseffecten, die in dit geval niet in de formulering van het probleem betrokken mogen worden, aldus ook Case Law T 2013, pag. 176–177 en 230 en T 936/96.
Voorts stelt AZ dat de uitvinding het wel in het octrooi (in paragraaf 0004) genoemde voordeel heeft dat de SR-formulering ongeveer dezelfde AUC heeft als de IR-formuleringen (‘while achieving blood plasma levels simular to those attained by administration smaller doses’). Bij de AUC (‘Area Under the Curve’) gaat het om de totale hoeveelheid werkzame stof in een bepaalde periode/ de beschikbaarheid in het bloed over de tijd opgeteld (het totale oppervlakte onder de curves in de hiervoor afgebeelde figuur). Nergens uit het octrooi blijkt — ook niet in paragraaf 0038 van het octrooi, waar over de AUC wordt gesproken — dat het verassend is dat de AUC bij toediening van een eenmalige SR-formulering ongeveer gelijk is aan de AUC bij meervoudige toediening van IR-formuleringen als beide binnen het therapeutisch bereik blijven. Kennelijk wordt ervan uitgegaan dat het vereiste dat de plasmaconcentratie binnen het therapeutisch bereik blijft, dit voor de vakman impliceert. Voorts wordt niet gesproken over bijzondere effecten of voordelen van het ongeveer gelijk zijn van de AUC, anders dan de voordelen die (al) voortvloeien uit de omstandigheid dat bij eenmalige toediening van de SR-formulering de plasmaconcentratie even lang binnen het therapeutische bereik blijft als bij meervoudige toediening van de IR-formulering, zoals weergegeven in figuur 2 in het octrooi. AZ heeft ter onderbouwing van haar stelling dat dit een onverwacht extra voordeel is slechts concreet gesteld dat dit een gevolg is van het hoge first pass metabolisme van quetiapine, waardoor bij een SR-formulering minder werkzame stof de bloedbaan bereikt dan bij IR-formuleringen; daarom ligt het niet in de lijn der verachtingen dat de AUC voor IR hetzelfde is als voor SR, aldus AZ. Nu deze stelling over de gevolgen van een hoog first pass metabolisme onjuist is (zie hierna ro. 33) gaat deze redenering niet op. Voor het overige heeft AZ niet voldoende onderbouwd waarom het ongeveer gelijk zijn van de AUC van een SR-formulering en IR-formuleringen onder voormelde condities bijzonder zou zijn. Naar het oordeel van het hof is er dan ook geen reden (ook) als technisch effect aan te merken en in de probleemstelling op te nemen dat de AUC ongeveer gelijk is.
20.
AZ stelt dat het probleem aldus moet worden omschreven: wat zou de vakman doen als hij de behandelmethode voor schizofrenie wilde verbeteren, althans het geneesmiddel quetiapine zou willen verbeteren. De eerste voorgestelde probleemstelling stuit al af op hetgeen hiervoor is overwogen over de CPA. De tweede op de formulering van de kenmerkende maatregelen in de conclusie(s) van het octrooi.
21.
Uitgaande van het bovenstaande dient de probleemstelling naar het oordeel van het hof omschreven te worden als: het ontwikkelen van een (oraal toe te dienen) formulering van quetiapine die een stabiel en gewenst plasma niveau bewerkstelligt (dat wil zeggen een plasmaniveau binnen het therapeutisch bereik) met een minder frequente toediening. AZ voert hier nog tegen aan dat het verkrijgen van een stabiel plasmaniveau op de indieningsdatum niet werd gezien als een probleem of wens van de vakman. Hieraan gaat het hof voorbij nu in het octrooi (in paragraaf 0002) een stabiel plasmaniveau als gewenst technisch effect van gereguleerde afgifte wordt genoemd. Voorts stelt AZ dat het verkrijgen van een stabieler plasmaniveau een resultaat van de uitvinding is en/of de probleemstelling een pointer naar de oplossing bevat en dus niet in de probleemstelling mag worden opgenomen en dat deze moet worden geherformuleerd. Het hof deelt dit oordeel niet. Dat een stabiel plasmaniveau het technisch effect van de onderscheidende maatregel is, is op zichzelf geen reden om deze niet in de probleemstelling op te nemen; integendeel. Het hof deelt niet het oordeel dat sprake is van een pointer naar de oplossing. Een stabiel plasmaniveau kan bovendien op meerdere manieren worden verkregen (zie hiervoor ro. 12).
22.
De vraag die thans derhalve beantwoord moet worden is of de uitvinding voor de hand lag voor de gemiddelde vakman, uitgaande van voormelde CPA en probleemstelling. Daarbij is de algemene vakkennis waarover die vakman op de prioriteitsdatum beschikte relevant. AZ betwist niet dat het de vakman ermee bekend was dat HPMC gebruikt kon worden om SR-formulering te maken van quetiapine en dat HPMC een veel daarvoor gebruikt middel was. Zij betwist echter dat de uitvinding voor de vakman voor de hand lag omdat hij, kort samengevat, niet gemotiveerd was om een SR-formulering van quetiapine te ontwikkelen en omdat hij geen redelijke verwachting van succes had/er contra-indicaties waren die hem van ontwikkeling zouden afhouden.
Motivatie
23.
Partijen twisten over het antwoord op de vragen of in casu, mede gelet op de toepassing van de PSA, moet worden onderzocht of de gemiddelde vakman gemotiveerd was om SR-formulering van quetiapine te ontwikkelen en of die motivatie een voorwaarde is om aan te nemen dat inventiviteit ontbreekt. AZ beantwoordt die vragen bevestigend, terwijl Sandoz die vragen ontkennend beantwoordt, stellende dat in de PSA de vakman wordt geacht er voortdurend op gericht te zijn het objectieve probleem op te lossen. AZ stelt dat die motivatie ontbrak, althans gering was omdat, kort gezegd:
- 1.
niet duidelijk was dat quetiapine (IR) in de praktijk effectief was, terwijl de risico's ervan onbekend waren (zie punt 17, onder a, b en c van de pleitnotities AZ) en
- 2.
er geen behoefte was aan een minder frequent toe te dienen formulering van quetiapine (punt 17, onder d, e, f, g, h, k en p (deels))
24.
Het hof is van oordeel dat het stellen van deze motivatievragen zich niet verdraagt met de CPA en de probleemstelling, waarvan naar het oordeel van het hof moet worden uitgegaan. Het probleem dat de vakman moet oplossen is immers een (oraal toe te dienen) formulering van quetiapine te ontwikkelen die een stabiel en gewenst plasma niveau bewerkstelligt met een minder frequente toediening. Bij de beantwoording van deze vraag kan niet betrokken worden óf de vakman gemotiveerd was een formulering van quetiapine te ontwikkelen en óf wel behoefte was aan een minder frequente toediening. Dat motivatie een rol kan spelen bij de beantwoording van de vraag of de vakman een bepaalde weg in zou slaan bij het zoeken naar een oplossing van het voorgelegde probleem is iets anders dan de vraag of de vakman überhaupt gemotiveerd was het aan hem voorgelegde probleem op te lossen. In zoverre slagen grieven 1 tot en met 4.
25.
Het hof is overigens van oordeel dat de vakman wel gemotiveerd was een formulering van quetiapine te ontwikkelen, nu er op de prioriteitsdatum voldoende aanwijzingen waren, op grond waarvan te verwachten was dat quetiapine effectief was (zou zijn), terwijl er geen reden was om aan te nemen dat er zodanige risico's waren dat de vakman daarvan om die reden zou afzien. Zulks op grond van de positieve rapportage over de werkzaamheid in onder meer S.R. Hirsch e.a. ICI 204,636: A New Atypical Antipsychotic Drug, British Journal of Psychiatry (A22) van mei 1996 (aldus punt 32 van de in zoverre niet betwiste uitspraak van de High Court of Justice van 22 maart 2012) en met name de persberichten van AZ en Eurand America Inc. van oktober 1995.
Het persbericht van AZ luidt:
‘Important results presented today from the first major Phase III clinical trial of ‘Seroquel’ confirm that this new ‘atypical’ antipsychotic being developed by Zeneca Pharmaceuticals should offer substantial benefits in the management of schizophrenia.
A convenient, twice daily dosing regimen of ‘Seroquel’ offers effective management of both the ‘positive and negative’ symptoms of schizophrenia and fewer of the disturbing side-effects associated with current antipsychotic drugs. Movement disorders (so-called extrapyramidal side effects) with ‘Seroquel’ have been no greater than those shown with placebo — a feature which helps to confirm its profile as an ‘atypical’ antipsychotic.’
Het gebruik van het woord ‘should’ kan niet (voldoende) afdoen aan de positieve verwachtingen van quetiapine.
26.
Het hof is bovendien overigens van oordeel dat er wel degelijk behoefte was aan een eenmaal daagse toediening. AZ stelt dat daaraan geen behoefte bestond omdat
- 1.
op de prioriteitsdatum een eendaagse toediening van de IR-formulering mogelijk werd geacht door Gefvert en Flischhacker;
- 2.
er voor wat betreft de therapietrouw tussen eenmaal daagse en tweemaal daagse toediening geen relevant verschil is.
Ad 1.
27.
Gefvert doet verslag van een onderzoek waarbij driemaal daags SEROQUEL was toegediend en suggereert dat een tweemaal daagse toediening (BID) van SEROQUEL (IR-formulering) succesvol kan zijn. Vermeld wordt dat er een grotere studie zal worden gedaan waarbij een tweemaal daagse toediening zal worden vergeleken met driemaal daagse toediening (TID). Uit het verslag blijkt dat de D2 bezetting tussen 12 en 26 uur daalt tot onder de 20% en daarmee onder het niveau om werkzaam te zijn, terwijl de halfwaarde tijd ongeveer 5,3 uur is. Beide bevindingen wijzen erop dat een eendaagse IR-formulering niet voldoende werkzaam zou zijn. Zie ook het export report van D.A. Rawlings, paragraaf 8 (S 16). Dat Gefvert opmerkt dat ‘once or twice daily dosing may therefore maintain sufficient 5Ht2/D2 recepter occupancy’ kan daaraan onvoldoende afdoen, nu dat niet wordt onderbouwd en de vakman zal begrijpen dat de vermelde gegevens die stelling in zoverre niet kunnen dragen dat een eendaagse toediening van een IR-formulering voldoende is. Dat wordt nog ondersteund door de aangekondigde vergelijkende studie tussen BID en TID. Overigens is ook de halfwaardetijd vermeld in de Practice Guideline for the Treatment with Schizophrenia van de American Psychiatric Association (1994) van 6,9 uur te kort voor een eendaagse toediening van een IR-formulering.
Ad 2.
28.
De vraag rijst in of en in hoeverre een eenmalige toediening ten opzichte van meerdaagse toediening de therapietrouw en/of het gebruiksgemak zou bevorderen. Naar het oordeel van de rechtbank zou de vakman weinig voordeel in therapietrouw verwachten van de overgang van tweemaal daags naar eenmaal daags; doseringsfrequentie zou niet het grootste probleem zijn bij de gebrekkige therapietrouw van psychiatrische patiënten (maar het vergeten medicatie te nemen en/of weigeren medicatie te nemen door gebrek aan ziekte-inzicht en/of vanwege de bijwerkingen), terwijl er ook andere manieren zouden zijn om het probleem van therapietrouw op te lossen, aldus de rechtbank. Als dit al zo zijn — de rechtbank somt een aantal andere mogelijkheden op die de therapietrouw kunnen vergroten (maar die alle duidelijke nadelen hebben) — dan doet het er niet aan af dat therapietrouw een probleem is en dat een eenmalige toediening een oplossing voor dat probleem is. De rechtbank heeft melding gemaakt van publicaties waarin gesproken wordt van een geringe verbetering, zoals RN. Greenberg (A10). Hiernaast staan publicaties waarin zonder meer is vermeld dat juist voor psychiatrische patiënten eenmalige toediening de therapietrouw bevordert, zoals het handboek van Aulton, pagina 315 (S 7).
Ook de klinische deskundigen van partijen, Van Praag (deskundige van Sandoz) in zijn eerste verklaring, in paragraaf 10 (S 17) en Kahn (deskundige van AZ) in zijn eerste verklaring onder 15c (A4) verklaren dat. Gelet hierop is het hof van oordeel dat een eendaagse toediening ten minste de therapietrouw in enige mate bevordert. Daarnaast is het hof van oordeel dat dit het gebruiksgemak voor de patiënt en voor de verpleging bevordert, zeker bij onwillige patiënten.
Redelijke verwachting van succes/contra-indicaties
29.
Het bovenstaande doet er naar het oordeel van het hof niet aan af dat in het kader van de vraag of de vakman het probleem zou hebben opgelost als geclaimd en of hij een redelijke verwachting van succes zou hebben, wel relevant is of (de vakman er redelijkerwijs van uit mocht gaan dat) de verkregen SR-formulering niet een mindere klinische werkzaamheid heeft dan de (meervoudige) IR-formulering. Juist bij chemische uitvindingen moet dit in aanmerking worden genomen (vergelijk Case Law 2013, p. 211 – 213).
30.
AZ heeft, afgezien van voormelde motivatieproblemen, de volgende elementen of problemen opgesomd (onder meer in punt 17 van de pleitnotities in hoger beroep, waarnaar het hof hieronder verwijst), die reden zouden zijn waarom de vakman geen redelijke verwachting van succes zou hebben en/of het probleem niet zou hebben opgelost als geclaimd:
- a.
niet gebleken is dat een SR-formulering tot minder bijwerkingen zou leiden (17, sub i en j);
- b.
een SR-formulering is niet geschikt voor de behandeling van een aanval (17, sub k);
- c.
bij een SR-formulering ontbreekt de vereiste D2-receptor bezetting (17, sub 1 en p) ;
- d.
een SR-formulering werkt niet door de lage biologische beschikbaarheid door het hoge first pass metabolisme/variabele kinetiek (17, sub m, p en q);
- e.
een SR-formulering werkt niet door een hoge eiwitbinding (17, sub n);
- f.
een SR-formulering is niet mogelijk door het (zeer sterk) pH afhankelijk oplosbaarheidsprofiel/ verschillen in oplossnelheid/ hoge oplosbaarheid (met dosedumping) (17, sub o);
- g.
vanwege grote omvang van de tablet bij een SR-formulering (17, sub r);
Ad a en b.
31.
Dit zijn problemen die in casu niet relevant zijn. De vakman, uitgaande van het op te lossen probleem, zoekt niet naar een formulering van quetiapine die minder bijwerkingen heeft of die geschikt is voor de behandeling van een aanval. De stellingen over (vermindering van) bijwerkingen zijn in dit geding ook aanvankelijk aan de orde gesteld door Sandoz als verweer tegen de — naar het oordeel van het hof niet relevante — stelling van AZ dat de vakman niet gemotiveerd zou zijn om een nieuwe formulering van quetiapine te maken. Overigens was de vakman op de prioriteitsdatum bekend met de resultaten van klinische studies onder meer beschreven in Gefvert, waaruit naar voren kwam dat er op het gebied van bijwerkingen nauwelijks problemen (bekend) waren.
Ad c.
32.
Dit probleem is niet genoemd in het octrooi, hetgeen een aanwijzing is dat de vakman dat toen niet als probleem zag. AZ heeft in eerste aanleg gesteld dat de vakman op de prioriteitsdatum dacht dat het vereiste plasmaniveau om voldoende D2-receptorbezetting te bereiken samenhing met het piek plasmaniveau. De minimaal vereiste D2-receptorbezetting was echter niet bekend. Het hof is van oordeel dat de vakman er op de prioriteitsdatum niet van uitging dat voor de atypische antipsychotica quetiapine en clozapine een D2-receptorbezetting van minimaal 60% noodzakelijk was, nu dat nergens uit bleek. Uit Gefvert bleek dat quetiapine in ieder geval werkzaam was bij een bezettingsgraad van 44%. De rechtbank heeft overwogen dat de gemiddelde vakman terughoudend zou zijn met het ontwikkelen van een formulering met quetiapine waarbij het maximale plasmaniveau lager zou uitkomen dan bij een IR-formulering, omdat hij er niet zonder meer van uit zou gaan dat dit geen invloed zou hebben op de bezetting van de D2-receptoren en (daarmee) de werkzaamheid van quetiapine. Hij zou onderkennen dat er een risico bestond dat de D2-receptorbezetting bij een vertraagde afgifteformulering van quetiapine, met een toch al relatief lage bezettingsgraad van 44% onder de (onbekende) kritische grens zou zakken, aldus de rechtbank. Het hof is van oordeel dat niet geconcludeerd kan worden dat 44% een relatief lage bezettingsgraad is, nu helemaal niet bekend was welke minimale D2-receptorbezetting vereist was voor werkzaamheid. Dat 44% relatief laag is wordt ten onrechte geconcludeerd op grond van een percentage waarvan slechts vaststaat dat het niet toepasselijk is. Bovendien is onvoldoende onderbouwd dat het piek-plasmaniveau in dit verband belangrijk is. Het hof is dan ook van oordeel dat onvoldoende (onderbouwd) is gesteld om aan te nemen dat de vakman geen redelijke verwachting van succes zou hebben vanwege een risico op onvoldoende D2-receptorbezetting.
Ad d.
33.
Quetiapine heeft een hoog first pass metabolisme, waardoor slechts een beperkte hoeveelheid stof de plaats van bestemming bereikt. De vraag rijst of de vakman hierin een probleem zou hebben gezien om tot een werkzame SR-formulering van quetiapine te komen in verband met een vermindering van de biologische beschikbaarheid. In het octrooi wordt dit probleem niet genoemd, terwijl P. Rue in zijn door Sandoz overgelegde rapporten onbetwist en met voorbeelden onderbouwd heeft gesteld dat er diverse SR-formuleringen van geneesmiddelen bekend zijn die een hoog first pass metabolisme vertonen. AZ stelt dat het daarbij niet steeds gaat om antipsychotica, maar dat is in dit verband, waar slechts de vraag voorligt of een hoog first pass metabolisme in de weg staat aan een SR-formulering, niet relevant. Voorts stelt AZ dat de SR-formulering van het psychofarmacum Remoxipride (genoemd in de publicatie van D. Tench e.a (S 28)) van de markt is gehaald, maar niet gesteld of gebleken is dat dit iets te maken heeft met het vermeende probleem van een hoog first pass metabolisme. De rechtbank heeft overwogen dat de vakman hierin wel een probleem zou zien omdat de werkzaamheid van een stof met een hoog first pass metabolisme wordt ontleend aan de hoeveelheid stof die het verzadigingsniveau overschrijdt en bij een vertraagde afgifte formulering het actief ingrediënt over een langere tijd in lagere concentraties wordt vrijgegeven, waardoor het verzadigingsniveau minder of niet wordt overtreden, zodat minder of geen geneesmiddel beschikbaar komt. Sandoz heeft gemotiveerd gesteld dat een hoog first pass metabolisme niet leidt tot een verschil tussen IR- en SR-formuleringen in de zin van biologische beschikbaarheid, omdat in de normale situatie (waarbij niet alle leverenzymen verzadigd zijn) sprake is van lineaire farmacokinetiek. Dat betekent dat een toename in de hoeveelheid aan de lever afgegeven geneesmiddel zal leiden tot een proportionele toename van de hoeveelheid gemetaboliseerd geneesmiddel. Steeds zal het percentage van de toegediende hoeveelheid hetzelfde zijn, tenzij zich de situatie voordoet dat er onvoldoende enzymen zijn om te metaboliseren (hetgeen met verzadiging wordt aangeduid), maar dat is niet de normale situatie. Een en ander valt af te leiden uit het abstract van Y.W.J. Wong e.a. ‘Multiple dose pharmacokinetics and dose proportionality study of Seroquel (ICI 204,636) in male schizophrenic patients’ (1995) (S 26) — hierna: Wong I. Daaruit blijkt dat zowel de Cmax als de AUC proportioneel steeg bij toediening van doses tussen 100 mg tot 375 mg, zodat sprake is van lineaire kinetiek. Tijdens het pleidooi in hoger beroep heeft de raadsman van AZ ook erkend dat de rechtbank met haar overwegingen over verzadiging ‘niet helemaal goed zit’. Evenzo voornoemde uitspraak van de High Court of Justice in rechtsoverweging 118. AZ stelt dat het erom gaat dat de vakman ziet dat het een probleem kan zijn. Het hof is van oordeel dat als de vakman al zou denken dat dit een probleem zou opleveren (wat niet erg aannemelijk is nu het octrooi dit ook niet als een probleem noemt), hij uit Wong I, dat behoort tot zijn algemene vakkennis en waarmee hij naar het oordeel van het hof bekend zou zijn, zou afleiden dat bij quetiapine sprake is van lineaire kinetiek. Vergelijk ook paragraaf 33 ev. van de tweede verklaring van dr. Rue in de Engelse procedure (S 25).
Ad e
34.
Quetiapine bindt zich in hoge mate aan eiwitten in het bloedplasma. Voor het passeren van de bloed-hersenbarrière (BBB), dat nodig is om de hersenen te bereiken (om biologisch beschikbaar te zijn) dient een geneesmiddel niet eiwitgebonden te zijn. De rechtbank heeft daaruit, in navolging van AZ, geconcludeerd dat een SR-formulering een risico inhield dat de klinische werkzaamheid onder een aanvaardbaar niveau kwam te liggen. Sandoz heeft er echter onbetwist op gewezen dat er geen enkele publicatie is waarin een hoge serumeiwitbinding als een specifiek probleem wordt gezien voor het ontwikkelen van een SR-formulering. Rue heeft in zijn eerste en tweede rapport (S 19 en 20; paragraven 26 respectievelijk 9) voorbeelden gegeven van SR-formuleringen van werkzame stoffen met een hoge serumeiwitbinding. Sandoz heeft onbetwist gesteld dat een hoge serumeiwitbinding geen probleem is omdat serumeiwitbinding een evenwicht in het bloed is en elke keer als ongebonden quetiapine de BBB over is het evenwicht hersteld moet worden en niet (meer) aan serumeiwit gebonden quetiapine vrijkomt, zodat ingeval van een SR-formulering dezelfde relatieve hoeveelheid ongebonden quetiapine beschikbaar zal zijn om de BBB te passeren als bij IR-formuleringen en de biologische beschikbaarheid dus ongeveer gelijk is. Sandoz geeft terecht aan dat Frijlink in zijn rapport, waarop AZ zich in dit verband beroept (A 24 , no 15), geen SR- en IR-formuleringen vergelijkt van dezelfde werkzame stof, maar slechts stelt dat sterke serumeiwitbinding de hoeveelheid beschikbare werkzame stof vermindert vergeleken met werkzame stoffen met een lagere serumeiwitbinding. Gelet op het bovenstaande acht het hof onvoldoende onderbouwd dat de vakman hierin een probleem zou zien, mede in aanmerking nemende dat het ook niet in het octrooi als een probleem wordt genoemd.
Ad f
35.
AZ stelt dat de oplosbaarheid van quetiapine varieert met de omgevings pH 1 — pH 7. AZ stelt dat er dat een groot verschil in oplosbaarheid zou zijn, namelijk met een factor 80 (bij lichaamstemperatuur) en dat door de extremen in wateroplosbaarheid de kans op dosdumping toeneemt doordat een te grote hoeveelheid geneesmiddel in de maag wordt gedumpt. Sandoz stelt dat de vakman hierin geen probleem zou zien omdat het verschil in oplosbaarheid bij deze pH waarden slechts een factor 8 is, veel kleiner dan de door AZ gesuggereerde factor 80. Voorts stelt Sandoz dat als de vakman hierin al een probleem zou zien, het tot de algemene vakkennis van de vakman behoort dat dit eenvoudig zou kunnen worden verholpen door het gebruiken van een buffer. Aldus blijkt bijvoorbeeld uit Dow (S 9; pagina 17, rechter kolom), waarin wordt gesteld: ‘it has also been reported that adding organic acids or buffers to the matrix can control the tablet's pH environment through the Gl tract’. Dit betekent dat de pH in de omgeving relatief constant wordt gehouden, waardoor pH afhankelijke oplosbaarheidsverschillen geen rol meer spelen. Dat dit onder meer tot gevolg kan hebben dat ‘drugs with poor atbility at certain pH ranges can be formulated to optimize bioavailability’ en het in casu juist gaat om het vraag of er niet een te hoge oplosbaarheid is, doet daar niet aan af. Er wordt hier door Dow slechts een voorbeeld gegeven van een van de (probleemoplossende) gevolgen van het toevoegen van een buffer, namelijk wanneer het gaat om een slecht oplosbare stof. Dat doet er niet aan af dat de pH neutralisatie eveneens plaatsvindt bij een (te) goed oplosbare stof. Evenzo dr. Rue in zijn verklaring in de Engelse procedure in paragraaf 19 -23 (S 25), die er bovendien op basis van de bevindingen van de deskundige van AZ, Moreton, op wijst dat geen sprake is van zodanige verschillen — maar slechts een achtvoudig verschil in oplosbaarheid tussen pH 1 en pH 7 — dat dit de vakman zorgen zou baren.
Ad g
36.
AZ stelt dat bij een eenmalige SR formulering een (te) grote tablet nodig zou zijn en dit een contra-indicatie zou zijn. Het hof is, met de rechtbank, van oordeel dat dit probleem eenvoudig kan worden opgelost door het formuleren van kleinere pil waarvan er twee per keer dienen te worden ingenomen. AZ stelt dat daarmee niet hetzelfde effect wordt bereikt als met één tablet, maar onderbouwt dat niet (voldoende). Sandoz heeft dit betwist en onbetwist gesteld dat quetiapine SR (800 mg) in de praktijk ook wordt gegeven met behulp van twee tabletten (zie de samenvatting van productkenmerken Seroquel XR van AZ (A 32B)). Het hof gaat aan dit vermeende probleem als onvoldoende onderbouwd voorbij. De voorwaardelijke incidentele grief van AZ, die zich richt tegen dit oordeel van de rechtbank, faalt derhalve.
37.
Nu de door AZ gestelde vermeende problemen voor een gemiddelde vakman niet in de weg stonden aan (een redelijke verwachting van succes bij) ontwikkeling van een SR-formulering van quetiapine met behulp van HPMC als geleermiddel, zou de vakman dit doen als hij geconfronteerd werd met voormelde probleemstelling en lag de uitvinding voor de vakman voor de hand, zodat conclusies 1 en 3 nietig zijn. De principale grieven 1 tot en met 7 en 9 slagen derhalve, terwijl grief 8 geen afzonderlijke behandeling behoeft.
38.
Het hof is van oordeel dat de volgconclusies 2 en 4 tot 20 niets inventiefs toevoegen. De onderbouwde stellingen van Sandoz dat en waarom deze conclusies ook niet inventief zijn als conclusie 1 (en 3) niet inventief is, wordt door AZ ook niet gemotiveerd betwist.
39.
In de conclusies zoals geformuleerd in de hulpverzoeken (A 20), wordt toegevoegd dat sprake is van een tabletvorm, dat sprake is van een ‘effective dosage amount’ van quetiapine, dat de vertraagde afgiftetijd tenminste 8 uur moet bedragen, dat quetiapine 35 tot 65% van formulering uitmaakt en dat de UAC ongeveer gelijk moet zijn aan de UAC van de IR-formulering. Hiermee wordt niets toegevoegd dat niet al in de oorspronkelijke conclusies door de vakman op basis van de beschrijving werd gelezen en/of daaraan inherent is (vergelijk wat de AUC betreft ro. 19). De conclusies volgens de hulpverzoeken zijn dus evenmin inventief. Bovendien leert het octrooi wat betreft de toevoeging dat er een ‘effective dosage amount’ van quetiapine moet zijn, niet (ook niet in paragraaf 0039) wat dat is en is het in wezen geen beperking.
40.
Het bestreden vonnis zal derhalve worden vernietigd, met veroordeling van AZ in de kosten van de procedure in eerste aanleg en hoger beroep. Partijen hebben zowel in eerste aanleg als in hoger beroep overeenstemming bereikt over de hoogte van de kosten, namelijk € 212.500,-- in eerste aanleg en € 125.000,-- voor het hoger beroep.
Beslissing
Het gerechtshof:
vernietigt het door de rechtbank Den Haag tussen partijen in conventie gewezen vonnis van 7 maart 2012
en opnieuw rechtdoende,
vernietigt van Nederlands deel van EP 0 907 364 B1;
veroordeelt AZ in de kosten van de procedure in eerst aanleg en hoger beroep, tot op heden aan de zijde van Sandoz begroot op € 212.500,-- voor de eerste aanleg en € 125.000,-- voor het hoger beroep;
verklaart de kostenveroordeling in dit arrest uitvoerbaar bij voorraad.
Dit arrest is gewezen door mrs. A.D. Kiers-Becking, M.Y Bonneur en C.J.J. van Loon; het is uitgesproken ter openbare terechtzitting van 10 juni 2014 in aanwezigheid van de griffier.