Daarin is per abuis niet het geactualiseerde proceskostenoverzicht van Novartis (EP16) opgenomen, ingekomen ter griffie op 11 maart 2022.
Rb. Den Haag, 21-06-2022, nr. C/09/625743 / KG ZA 22-195
Uitspraak 21‑06‑2022
Inhoudsindicatie
ctrooirecht. Kort geding. Gerede kans dat octrooi in een oppositie procedure of een bodemprocedure wordt herroepen of vernietigd vanwege een gebrek aan inventiviteit. Toepassing HR Boehringer / Kirin Amgen.
Partij(en)
vonnis
RECHTBANK DEN HAAG
Team handel
zaaknummer / rolnummer: C/09/625743 / KG ZA 22-195
Vonnis in kort geding van 21 juni 2022
in de zaak van
de rechtspersoon naar vreemd recht
NOVARTIS AG,
te Bazel, Zwitserland,
eiseres,
advocaat mr. R.M. Kleemans te Amsterdam,
tegen
1. MYLAN B.V.,
te Amstelveen,
2. de rechtspersoon naar vreemd recht
MYLAN IRELAND LIMITED,
te Dublin, Ierland,
gedaagden,
advocaat mr. J.J.E. Bremer te Den Haag.
Partijen zullen hierna Novartis en Mylan c.s. genoemd worden en gedaagden ook afzonderlijk Mylan BV en Mylan Ltd. De zaak is voor Novartis inhoudelijk behandeld door mr. Kleemans voornoemd, mr. J.D. Drok en mr. A.F. Tadema, advocaten te Amsterdam en voor Mylan c.s. door mr. Bremer voornoemd, mr. M.H.J. van den Horst en mr. A.H. van Duijn, advocaten te Den Haag
1. De procedure
1.1.
Het verloop van de procedure blijkt uit:
- het tussenvonnis van 22 maart 2022 en de daarin genoemde gedingstukken;
- -
het tussenvonnis van 23 maart 2022 waarin hoger beroep is opengesteld tegen de eindbeslissing in voorlopige voorziening in het tussenvonnis van 22 maart 2022;
- -
de akte van Mylan c.s., ingekomen ter griffie op 12 april 2022, met producties GP18 en GP19;
- -
de antwoordakte van Novartis, ingekomen ter griffie op 2 mei 2022, met producties EP17 tot en met EP22;
- -
de akte overlegging productie GP16.2 van Mylan c.s., ingekomen ter griffie op 16 mei 2022, met productie GP16.2 (een geactualiseerd proceskostenoverzicht);
- -
de mondelinge behandeling van 17 mei 2022 en de ter gelegenheid daarvan overgelegde pleitnota’s van Mylan c.s. en Novartis. Van de pleitnota van Mylan c.s. zijn de voetnoten (waarvan er een aantal niet uitsluitend een bronvermelding bevatten) niet gepleit, van de nummers 8, 10, 19 en 59 zijn de citaten niet gepleit en van nummer 21 is enkel het onderstreepte deel van het citaat gepleit. Daarnaast is nummer 5 niet gepleit. Van de pleitnota van Novartis zijn de voetnoten (waarvan er eveneens een aantal niet uitsluitend een bronvermelding bevatten) niet gepleit en daarnaast zijn de nummers 4, 38, 60 en 61 niet gepleit. Op die delen is geen acht geslagen.
1.2.
Mylan c.s. heeft op 29 april 2022 een akte overlegging productie overgelegd, met productie GP20. Novartis heeft bezwaar gemaakt tegen overlegging van deze akte/productie. Nadat partijen over en weer per e-mail hun standpunten ter zake bekend hebben gemaakt, heeft de voorzieningenrechter bij e-mail van 3 mei 2022 de akte en productie GP20 geweigerd.
1.3.
Op 13 juni 2022 hebben partijen gezamenlijk de schriftelijke uitwerking van de reasons for the decision van de Technische Kamer van Beroep (TKB) van het Europees Octrooibureau (EOB) aan de voorzieningenrechter toegezonden. Vonnis is naar aanleiding daarvan nader bepaald op heden.
1.4.
Op 14 juni heeft Mylan c.s. verzocht nog een akte te mogen nemen over de reasons for the decision van de TKB. Novartis heeft bezwaar gemaakt tegen dat verzoek. Het verzoek is door de voorzieningenrechter geweigerd omdat de mondelinge behandeling in het kort geding al had plaatsgevonden. Vervolgens hebben beide partijen nog e-mails gestuurd aan de voorzieningenrechter, waarin zij probeerden op die wijze nog standpunten in te nemen. Om de reden waarom de akte is geweigerd, heeft de voorzieningenrechter ook geen acht geslagen op die brieven.
2. De feiten
2.1.
Met verwijzing naar 2.1 tot en met 2.3 van het tussenvonnis van 22 maart 2022 (hierna: het tussenvonnis), herhaalt de voorzieningenrechter voor de leesbaarheid het volgende. De Novartis-groep produceert en verhandelt onder andere het geneesmiddel met de werkzame stof fingolimod (ook aangeduid als FTY720) voor de behandeling van relapsing-remitting multiple sclerose (hierna: RRMS). Novartis heeft op 16 juli 2015 een Europese octrooiaanvraag ingediend onder nummer EP 2 959 894 (hierna wederom: aanvraag EP 894). Aanvraag EP 894 is een afgesplitste aanvraag, waarvan de grootmoederaanvraag (met nummer EP 2 037 906) op 25 juni 2007 is ingediend, die op haar beurt prioriteit inroept van GB0612721, gedateerd 27 juni 2006. De TKB van het EOB heeft tijdens de zitting van 8 februari 2022 beslist dat aan Novartis een octrooi verleend zal worden op basis van aanvraag EP 894 en heeft de zaak terugverwezen naar de EDdie het octrooi dient te verlenen op basis van de volgende (enige) conclusie van het hoofdverzoek:
A S1P receptor modulator for use in the treatment of relapsing-remitting multiple sclerosis, at a daily dosage of 0.5 mg p.o., wherein said S1P receptor modulator is 2-amino-2-[2-(4-octylphenyl)ethyl]propane-1,3-diol in free form or in a pharmaceutically acceptable salt form.
2.2.
In de (onbestreden) Nederlandse vertaling luidt deze conclusie als volgt:
S1P-receptor-modulator voor toepassing bij de behandeling van relapsing-remitting multiple sclerose, in een dagelijkse dosis van 0,5 mg p.o., waarbij de S1P-receptor-modulator 2-amino-2-[2-(4-octylfenyl)ethyl]propaan-1,3-diol in vrije vorm of in een farmaceutisch aanvaardbare zoutvorm is.
2.3.
In aanvulling op de feiten die in het tussenvonnis zijn opgenomen, zijn de volgende feiten relevant voor het onderhavige geschil.
2.4.
Fingolimod is een S1P-receptor. S1P-receptoren zijn gebonden aan lymfocyten. Een lymfocyt is een bepaald type witte bloedcel dat deel uitmaakt van het immuunsysteem. Door de binding van fingolimod (middels de S1P-receptoren) aan de lymfocyten worden deze reversibel vastgehouden in de lymfeklier, waardoor deze lymfocyten niet in het bloed en in het centrale zenuwstelsel kunnen circuleren, wat neuroinflammatie en myeline beschadiging vermindert in de hersenen en het ruggenmerg. Door dit werkingsmechanisme kan fingolimod worden toegepast in de behandeling van een specifieke vorm van multiple
sclerose (MS).
2.5.
De navolgende publicaties behoren voor aanvraag EP 894 tot de stand van de techniek.
2.5.1.
Media release Novartis, “Phase II data for FTY720 shows sustained efficacy and good tolerability over 18 months in patients with relapsing multiple sclerosis (MS)”, 6 april 2006 (hierna: media release Novartis). Media release Novartis is in de verleningsprocedure voor het EOB D10 genoemd en vormde voor de TKB de dichtstbijzijnde stand van de techniek. Daarin is – onder meer – opgenomen:
Phase II study design
The results are from a large Phase II study conducted at 32 centers in 11 countries (Europe and Canada). In the initial, placebo-controlled part of this study, 281 patients were randomized in equal numbers to receive either placebo, 1.25 mg or 5 mg of FTY720 orally once-daily for six months. The study evaluated the effect of FTY720 on disease activity as measured by MRI and clinical relapses as well as its tolerability and safety. After six months, patients had the option to enter the extension phase evaluating the longer-term effects. Patients in the placebo group were re-randomized to receive either 1.25 mg or 5 mg, while patients already on FTY720 continued their originally-assigned treatment. Having completed the 12 month time point, the 5 mg dose arm was discontinued and patients previously receiving this dose were continuing in the study on a dose of 1.25mg.
Analysis of the 24-month data is expected to be presented at a key neurological congress in the second half of 2006.
Phase III study program
Novartis has initiated its first Phase III pivotal study called "FREEDOMS" (Fingolimod Research Evaluating Effects of Daily Oral therapy in Multiple Sclerosis). The 24-month, randomized, double-blind, placebo-controlled FREEDOMS study will include more than 1,000 patients with relapsing-remitting MS between age 18-55. Study participants will be equally randomized to either receive either 1.25 mg or 0.5 mg of FTY720 or placebo once daily for up to 24 months.
This study has begun enrolling patients in several European countries. Novartis is currently in discussions with the US Food and Drug Administration (FDA) on Phase III initiation in the US.
2.5.2.
Presentatie Novartis, “FTY 720 - A novel oral agent for Multiple Sclerosis (MS) – Results from a 6 month proof of concept study”, gepubliceerd voor 16 maart 2006 (hierna: presentatie Novartis). In de verleningsprocedure voor het EOB is dit document TPO-D1 genoemd. Hieronder is dia 26 van deze presentatie opgenomen:

2.5.3.
Journal of Neurology abstracts, P569, “Design of a randomised, placebo-controlled study of oral fingolimod (FTY720) in relapsing-remitting multiple sclerosis”, L. Kappos et al, mei 2006 (hierna: Kappos). Daarin is opgenomen:

2.5.4.
Pharmacodynamics, pharmacokinetics, and safety of Multiple Doses of FTY720 in stable renal transplant patients: a multicenter, randomized, placebo-controlled, phase I study, Kahan e.a., 2003 (hierna: Kahan). Kahan openbaart onderzoek naar doseringen van 0.125, 0.25, 0.5, 1.0, 2.5 en 5.0 mg per dag bij transplantatie-patiënten. De 0.5 mg per dag dosering leidde tot een lymfocytenreductie van ongeveer 50% in deze studie.
2.5.5.
Peripheral blood FTY720 pharmacokinetic/pharmacodynamic (PK/PD) modelling in renal transplanted recipients, Park e.a., 2003 (hierna: Park 2003). Park 2003 heeft eveneens betrekking op transplantatie-patiënten. Het openbaart een studie naar doseringen van onder andere 0.5 mg per dag. Uit de resultaten in Park 2003 komt naar voren dat de 0.5 mg per dag dosering leidde tot een lymfocytenreductie van 44%, wat volgens dat document betekende dat die dosering onvoldoende therapeutisch effect had.
2.5.6.
Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting
experimental autoimmune encephalitis in SJL mice, Webb e.a., Journal of Neuroimmunology 153 (2004) (Hierna: Webb), waarin is opgenomen:

en onder ‘discussion’:


2.5.7.
Pharmacokinetic/pharmacodynamic relationships of FTY720 in kidney transplant recipients, Park e.a., 2005 (hierna: Park 2005). Park 2005 openbaart ook een lymfocytenreductie van ongeveer 44% bij een dosering van 0.5 mg per dag. Daarin is voorts vermeld:
Inter and intra-individual variability of percent reduction of lymphocyte counts decreases with increasing FTY720 doses, perhaps due to achievement of maxima/pharmacodynamic effect.
Park bevat de onderstaande figuren:
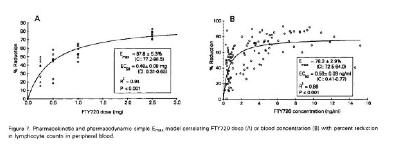
2.5.8.
FTY720 in multiple sclerosis: the emerging evidence of its therapeutic value, Thomson, gepubliceerd in maart 2006 (hierna: Thomson). In dit overzichts-artikel over fingolimod is vermeld (p.162-164):

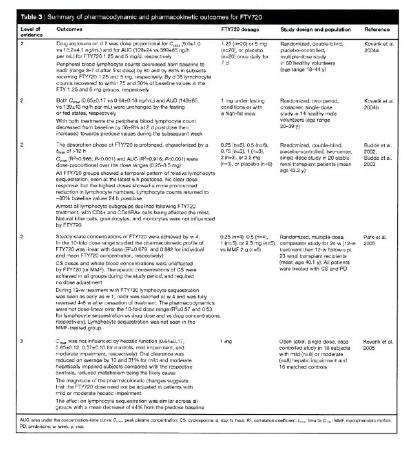

2.6.
In de Decision to refuse a European Patent application (waarin aanvraag EP 894 door de ED is geweigerd) van 19 november 2020 (hierna: de ED Decision) is – voor zover hier van belang – het volgende opgenomen:
I. Summary of Facts and Submissions
(…)
14 Third party observations (…)
These observations primarily concerned Article 83 EPC and the extent to which the subject matter of the claims did actually provide an effective treatment tor relapsing-remitting multiple sclerosis. Included in these observations were also comments relating to clarity (Article 84 EPC), extension of subject matter (Article 123(2) EPC), novelty (Article 54 EPC) and inventive step (Article 56 EPC). During the course of the written procedure, the applicant did respond to these observations and provided a series of experimental data supporting the case, particularly data providing a foundation tor sufficiency of disclosure under Article 83 EPC.
(…)
II. Decision
(…)
16.2
Novelty (Article 54 EPC)
16.2.1
The examining division is of the opinion that the subject matter of claim 1 of the main request lacks novelty over document D10. Document D10 (also labelled as TM1 in the file) is a media release from Novartis and a copy is annexed to this decision.
16.2.2
Claim 1 of the main request reads as follows:
A S1P receptor modulator for use in the treatment of relapsing-remitting multiple sclerosis, at a daily dosage of 0.5 mg p.o., wherein said S1P receptor modulator is 2-amino-2-[2-(4-octylphenyl) ethyl]propane-1,3-diol in free form or in a pharmaceutically acceptable salt form.
16.2.3
It is the opinion of the examining division that all of these features are directly and unambiguously disclosed in the above-mentioned prior art document D10.
2.7.
In de Minutes of the oral proceedings van de TKB van 8 februari 2022 (hierna: de Minutes) is – voor zover hier van belang – het volgende opgenomen:
The appellant confirmed its opening requests as follows:
- -
The appellant requested that the decision under appeal be set aside and that the case be remitted to the examining division with an order to grant a patent on the basis of the single claim of the main request underlying the impugned decision and a corresponding description of 15 pages filed on 7 January 2022.
- -
(…)
- -
The appellant further requested that
- the case not be remitted to the examining division for further prosecution
- the case not be stayed until a decision has been reached in the pending referral G 2/21
- the third-party observations filed on 27 April 2021, 2 November 2021, 17 November 2021, 9 December 2021 and 23 December 2021 not be admitted into the proceedings
- document D39 submitted with the third-party observations dated 27 April 2021 not be admitted into the proceedings;
- document TPO-D1 submitted with the third-party observations dated 9 December 2021 not be admitted into the proceedings
- documents D40 and D41 submitted with the third-party observations dated 23 December 2021 not be admitted into the proceedings
- appellant's submissions dated 7 January 2022 and documents D42 to D45 filed therewith be admitted into the proceedings
- document D46 filed on 7 January 2022 be admitted into the proceedings, should the board decide to admit document D39 into the proceedings.
(…)
The Chair then closed the debate and gave the following decision of the board:
1. The decision under appeal is set aside.
2. The case is remitted to the examining division with the order to grant a patent on the basis of the single claim of the main request filed on 18 November 2019 underlying the impugned decision and resubmitted with the statement of grounds of appeal, and a description to be adapted thereto.
2.8.
In de schriftelijke uitwerking van de beslissing van de TKB, die op 3 juni 2022 is gepubliceerd (hierna: de Decision), is opgenomen:
3. Admittance of the third-party observations received on 27 April 2021, 2 November 2021, 17 November 2021, 9 December 2021, 23 December 2021 and 18 January 2022
within the meaning of Article 115 EPC
3.1
All of these submissions have been received after the filing of the statement of grounds of appeal.
3.2
With this statement, the appellant had requested as the main request that the decision under appeal be set aside and that a patent be granted on the basis of the single claim of the main request underlying the impugned decision.
3.3
The board notes that this claim request had already been filed on 18 November 2019, i.e. almost one year before oral proceedings took place before the examining division.
3.4
In view of the foregoing, the board considers that the third-party observations received on 27 April 2021, 2 November 2021, 17 November 2021, 9 December 2021, 23 December 2021 and 18 January 2022 could and should have been filed during the examination proceedings. As a consequence, the board decided not to take these observations into account.
(…)
5.4
In the case at hand, the appellant submitted that the following findings on fingolimod were disclosed in the prior art.
( a) The lowest effective dose of fingolimod reported in animal models of EAE was 0.1 mg/kg/day p.o. (see document D14, page 17, left-hand column, last paragraph, first sentence).
( b) From these models, a threshold of about 70%depletion of peripheral lymphocytes was believed to be required to see any efficacy (see document D28, paragraph bridging pages 118 and 119).
( c) The claimed dose of 0.5 mg of fingolimod had been shown not to meet this threshold in stable transplant patients (see document D27, Figure 1) and acute transplant patients (see document D26, Figure 7A)
( d) Pharmacokinetic and pharmacodynamic outcomes following single- or multiple-dose administration of fingolimod in transplantation patients could be extrapolated to patients with MS (see document D23, page 162, right-hand column, second full paragraph).
5.5
On the basis of these facts, the board concludes that the prior art does not support the suitability of the claimed dosage regimen for the claimed therapeutic application.
(…)
5.27
As a consequence, the board judges the experimental data reported in the application in the context of the EAE study to be sufficient for establishing the initial plausibility of the therapeutic benefit of the claimed dosage regimen in human RRMS patients. Initial plausibility being given, the outcome of referral G 2/21, currently pending before the Enlarged Board of Appeal, is not decisive for the decision in the present case (see point 5.6 above).
(…)
7. Inventive step (Article 56 EPC)
The closest prior art
7.1
In agreement with the appellant, the board considers document Dl0's disclosure concerning the successful phase II trial on RRMS patients treated with fingolimod at an oral daily dose of 1.25 mg (see point 6.2(a) above) to be a suitable starting point for assessing inventive step of the claimed subject-matter.
7.2
The subject-matter of claim 1 differs from this disclosure in that fingolimod is administered at an oral daily dose of 0.5 mg.
Objective technical problem and solution
7.3
To formulate the objective technical problem, it is necessary to establish the technical effect(s) achieved by the aforementioned distinguishing feature.
7.4
For the reasons given above regarding sufficiency of disclosure, the board is satisfied that the claimed fingolimod dosage regimen provides an effective therapeutic treatment of RRMS.
7.5
Hence, starting from the closest prior art defined above, the objective technical problem is the provision of further means to effectively treat RRMS.
7.6
The proposed solution to this problem is the fingolimod dosage regimen recited in claim 1.
(…)
Obviousness
7.8
As set out in point 6.2 above, document Dl0's disclosure of the successful phase II trial with an oral daily dose of 1.25 mg fingolimod is immediately followed by the announcement of a phase III study using oral daily dose of 0.5 mg and 1.25 mg of fingolimod.
7.9
In the board's judgement, this announcement would have provided the skilled person with a reasonable expectation of solving the objective technical problem with fingolimod at an oral daily dose of 0.5 mg, unless a teaching in the prior art would have dissuaded the skilled person from considering this dosage regimen as a solution to the technical problem posed.
7.10
In the case at hand, the board is satisfied that the prior art teaches away from the claimed invention. As set out in point 5.4(b) above, the skilled person would have inferred from document D28 that a threshold of lymphocyte reduction of at least 70% was required for a therapeutic treatment of RRMS. Moreover, the board agrees with the appellant's position that the teachings of documents D26 and D27 taken in combination with the teaching of document D23 would have led the skilled person to conclude that an oral daily dose of 0.5 mg fingolimod would be insufficient for reaching this threshold and hence would not be therapeutically effective in the treatment of RRMS (see points 5.4(c) and 5.4 (d) above).
7.11
In light of these particular circumstances, the board finds that the announcement of the phase III trial in document Dl0 would have given the skilled person hope of success but not a reasonable expectation of it.
7.12
A mere hope of success does not suffice as motivation to render the claimed subject-matter obvious. As a consequence, the subject-matter of claim 1 involves an inventive step starting from Dl0's disclosure of the successful phase II trial with an oral daily dose of 1.25 mg fingolimod.
7.13
For the sake of completeness, the board observes that the same considerations apply when starting the assessment of inventive step from the announcement of the phase III trial in document Dl0 taken as the closest prior art (see point 6.2 above).
2.9.
Novartis heeft bij het EOB een verklaring van een deskundige ingebracht, prof. P. van der Graaf, die daarin onder andere het volgende heeft verklaard:

en

3. Het geschil
3.1.
Voor een weergave van het geschil en de stellingen en verweren van partijen in de onderhavige zaak verwijst de voorzieningenrechter naar paragraaf 3 van het tussenvonnis. Voor de leesbaarheid neemt de voorzieningenrechter hieronder nogmaals de subsidiaire vorderingen onder B op (die thans nog ter beoordeling voorliggen) en de nevenvorderingen die daarmee samenhangen:
Novartis vordert dat de voorzieningenrechter
(…)
bij vonnis, voor zover mogelijk uitvoerbaar bij voorraad:
(…)
subsidiair:
3. Mylan c.s. zal verbieden om vanaf de verlening van EP 894 tot aan het verlopen van EP 894 op 24 juni 2027 in Nederland inbreuk te maken op het Nederlandse deel van EP 894, en in het bijzonder fingolimod Mylan te vervaardigen, in het verkeer te brengen, verder te verkopen, af te leveren of anderszins te verhandelen, dan wel voor een of ander aan te bieden, in te voeren of in voorraad te hebben, dan wel daarbij op onrechtmatige wijze betrokken te zijn; en
4. Mylan c.s. zal bevelen om ieder product dat na de verlening van EP 894 inbreuk zal maken op het Nederlandse deel van EP 894 op eigen kosten in Nederland terug te halen; en
5. Mylan c.s. zal gebieden om alle afnemers van fingolimod Mylan uiterlijk bij de koop van fingolimod Mylan op een moment tussen 22 maart 2022 en het moment dat het octrooi wordt verleend, erover te informeren dat er binnenkort een octrooi zal worden verleend waarop fingolimod Mylan inbreuk maakt en dat dat betekent dat Mylan weliswaar op dit moment gerechtigd is om fingolimod Mylan te verkopen maar dat zij dat niet langer zal zijn op het moment dat het octrooi verleend wordt; en
6. Mylan c.s. zal gebieden dat zij op het moment dat zij door Novartis op de hoogte wordt gesteld dat EP 894 is verleend, onmiddellijk alle afnemers van fingolimod Mylan in Nederland schriftelijk bij brief informeert dat fingolimod Mylan niet langer verhandeld mag worden en door Mylan op eigen kosten zal worden teruggenomen;
7. Mylan c.s. zal gebieden om de verkoop van fingolimod Mylan aan alle afnemers nauwkeurig bij te houden zodat achteraf kan worden vastgesteld hoeveel van deze producten in Nederland zijn verkocht, en aan wie, en wat de verkoopprijs is geweest, inclusief eventuele kortingen, en wat de totale omzet is die daarmee is behaald, en welke winst daarmee is behaald, geverifieerd door een register accountant, en al deze informatie in ieder geval binnen dertig dagen nadat Novartis Mylan c.s. erover heeft geïnformeerd dat het octrooi is verleend, aan Novartis te verstrekken;
8. zal bepalen dat Mylan c.s. een onmiddellijk opeisbare dwangsom verbeurt van € 100.000,- per dag, dat zij in gebreke is ter zake van de onder 3) en 4) te geven verboden/bevelen, dat Mylan c.s. een onmiddellijk opeisbare dwangsom verbeurt van € 1.000,- per dag dat zij in gebreke is ter zake van de onder 5) en 6) te geven verboden/bevelen, en dat Mylan c.s. een onmiddellijk opeisbare dwangsom verbeurt van € 10.000,- per dag dat zij in gebreke is ter zake van het onder 7) te geven verbod/bevel;
alsmede zowel onder A en B, zowel primair als subsidiair:
I. de termijn voor het instellen van een eis in de hoofdzaak op grond van artikel 1019i Rvzal bepalen op zes maanden na het in dezen te wijzen vonnis; en
Mylan c.s. zal veroordelen in de volledige kosten van deze procedure conform artikel 1019h Rv.
4. De verdere beoordeling
Nietigheid van de (te verlenen) aanvraag EP 894
4.1.
De voorzieningenrechter dient, gelet op het nietigheidsverweer door Mylan c.s. tegen aanvraag EP 894, te beoordelen of er een gerede (dat is een serieuze, niet te verwaarlozen) kans aanwezig is dat aanvraag EP 894 (met de conclusie beschreven in 2.6 waarmee het octrooi zal worden verleend) in oppositie wordt herroepen of dat het Nederlandse deel van aanvraag EP 894 in een bodemprocedure nietig wordt bevonden.
Afstemmingsregel toepassen?
4.2.
Novartis heeft zich op het standpunt gesteld dat de voorzieningenrechter haar voorlopig oordeel over de geldigheid van het (te verlenen) octrooi in de onderhavige procedure dient af te stemmen op de beslissing van de TKB in de verleningsprocedure, die aanvraag EP 894 geldig acht en de ED heeft opgedragen over te gaan tot verlening. Gelet op die beslissing is aannemelijk dat er geen gerede kans is dat aanvraag EP 894 wordt herroepen of vernietigd, aldus Novartis.
4.3.
De afstemmingsregel brengt tot uitdrukking dat binnen het Nederlandse systeem van rechtsvordering de rechter in de bodemprocedure, waarin finaal wordt beslist, het primaat heeft boven de voorzieningenrechter die voorlopig oordeelt in kort geding. In het arrest Boehringer / Kirin Amgen heeft de Hoge Raad geoordeeld dat het het Hof in die zaak vrij had gestaan om de afstemmingsregel overeenkomstig toe te passen op een beslissing van de OD, omdat het Hof die beslissing op één lijn mocht stellen met een in een bodemprocedure gegeven oordeel waarvoor de afstemmingsregel geldt.De Hoge Raad schrijft deze overeenkomstige toepassing van de afstemmingsregel niet voor, maar plaatst zijn oordeel in het algemene kader van de hierboven al beschreven toets of er een ‘gerede kans’ bestaat dat in hoger beroep, anders dan in eerste aanleg, de oppositie wel gegrond zou worden bevonden. Enerzijds is het enkele herhalen van al door de OD beoordeelde en verworpen argumenten en de enkele mogelijkheid van een andere beoordeling van die argumenten door de TKB of de bodemrechter, volgens de Hoge Raad onvoldoende om de gerede kans aanwezig te achten. Het zal daarvoor ten minste aannemelijk moeten zijn dat de TKB of de Nederlandse bodemrechter anders zal oordelen dan de OD.Anderzijds is voor het oordeel dat een gerede kans bestaat dat de TKB het octrooi zal herroepen of de bodemrechter het zal vernietigen, niet slechts plaats in het geval dat aannemelijk wordt gemaakt dat de OD zodanige fouten heeft gemaakt, dan wel op zulk belangrijk materiaal geen acht heeft geslagen (of heeft kunnen slaan), dat te verwachten is dat, waren die fouten niet gemaakt of het materiaal wel in de beschouwingen betrokken, anders was beslist. Een kennelijke misslag of novum wordt door de Hoge Raad niet vereist. Deze criteria spelen dus een rol bij de beoordeling, maar in beginsel dient de voorzieningenrechter derhalve onverminderd de in 4.1 beschreven toets toe te passen.
4.4.
In deze zaak is er te minder aanleiding om af te stemmen op het oordeel van de TKB om de volgende redenen. Het arrest Boehringer / Kirin Amgen had betrekking op een beslissing van de OD in een oppositieprocedure. In de onderhavige zaak vraagt Novartis om de afstemmingsregel toe te passen op de beslissing van de TKB in de verleningsprocedure. Anders dan Mylan c.s. betoogt, kan de verleningsprocedure bij de TKB in beroep van een beslissing om niet te verlenen, niet gelijk worden gesteld aan een verstekprocedure zoals we die in Nederland kennen. De TKB beoordeelt de aanvrage ex officio inhoudelijk en neemt daarbij ook third party opinions mee bij de beoordeling. Die toetsing is minder marginaal dan de verstektoets waarbij de rechter alleen beoordeelt of een vordering niet onrechtmatig of ongegrond voorkomt. De verleningsprocedure kan echter geenszins worden gezien als een volwaardige procedure op tegenspraak. Derden kunnen in de verlenings-procedure (mits tijdig ingediend) een opinie geven en documenten aandragen die in hun ogen in de weg staan aan verlening, maar zij kunnen niet bij de mondelinge behandeling worden gehoord of daar weerwoord geven. Van een voldragen debat is daarom geen sprake. Daarin verschilt de verleningsprocedure dus van de oppositie-procedure bij het EOB, waarin wel tegenspraak mogelijk is.
Gerede kans dat octrooi niet in stand blijft?
4.5.
Bij de beoordeling van de gerede kans dat aanvraag EP 894 uiteindelijk wordt herroepen of vernietigd, weegt de voorzieningenrechter ten voordele van Novartis met name mee dat door Mylan c.s. min of meer dezelfde stand van de techniek wordt aangedragen als al in de verleningsprocedure aan de orde is gesteld. De TKB is bij de beoordeling van de nieuwheid en inventiviteit uitgegaan van media release Novartis als meest nabije stand van de techniek, een document dat Mylan c.s. ook aan haar nietigheidsverweer ten grondslag legt. Dat document bevat ook bijna alle kenmerken die worden geopenbaard in presentatie Novartis, het document waarvan Mylan c.s. stelt dat dat de dichtstbijzijnde stand van de techniek is. Desalniettemin is de voorzieningenrechter voorshands van oordeel dat er een gerede kans is dat het octrooi niet in stand blijft in oppositie of dat het Nederlandse deel zal worden vernietigd in een nietigheidsprocedure. Daarvoor is het volgende redengevend.
4.5.1.
De relevante vakman voor dit octrooi is een team van een neuroloog met bijzondere kennis van de behandeling van MS en een pharmacoloog met kennis van pharmacokinetiek en -dynamiek, de ontwikkeling van MS geneesmiddelen en de in studies daarvoor gebruikte diermodellen. Partijen gaan daar in de door hen aangedragen deskundigenverklaringen ook (impliciet) vanuit.
4.5.2.
Uitgaande van media release Novartis (zie 2.5.1) is het enige kenmerk waarin de conclusie van EP 894 verschilt van deze stand van de techniek, het therapeutisch effect van de dosering 0,5 mg per dag (p.o.) voor de indicatie RRMS. Media release Novartis openbaart immers dat dagelijkse orale doseringen van 1.25 mg en 5 mg voor de indicatie RRMS therapeutische werking hebben en dat die werking bij beide doseringen gelijk is. Uitgaande van media release Novartis is het voor de vakman op te lossen technisch probleem het vinden van een gelijkwaardige behandeling van RRMS. De voorzieningenrechter volgt bij deze probleemstelling de TKB (zie 2.8, overweging 7.5: the objective technical problem is the provision of further means to effectively treat RRMS).
4.5.3.
Het behoort tot de algemene vakkennis van de vakman dat lagere doseringen verminderde bijwerkingen geven bij een groter deel van de patiëntenpopulatie, dus hij/zij zal altijd streven naar het vinden van een lagere dosering die anderzijds nog wel (voldoende effectieve) therapeutische werking heeft. Omdat media release Novartis openbaart dat er geen verschil is in therapeutische werking tussen de doseringen 1.25 mg en 5 mg per dag, zal de vakman inzien dat er bij die twee doseringen sprake is van een plateau. Daaruit leidt hij/zij af dat er een zekere doseringsrange lager dan 1.25 mg per dag moet zijn die ook voldoende therapeutische werking zal hebben, maar waarschijnlijk minder bijwerkingen. De vakman zal dus een succesverwachting hebben dat er een dosering is lager dan 1.25 mg per dag p.o. die gelijkwaardig is, omdat die dosering ook een effectieve therapie biedt met waarschijnlijk minder bijwerkingen.
4.5.4.
In media release Novartis leest de vakman dat er een Phase III klinische studie wordt gedaan naar een 0.5 mg per dag dosering, naast een dosering van 1.25 mg en placebo, naar aanleiding van Phase II studies met 1.25 mg en 5 mg per dag (p.o.). Phase III studies zijn langdurige klinische studies in een grote patiëntengroep, gericht op het verkrijgen van aanvullende informatie over klinische werking en statistisch bewijs van effectiviteit en veiligheid, dat nodig is voor registratie van een geneesmiddel. De eigen deskundige van Novartis Van der Graaf schrijft over Phase III studies in een verklaring: ‘There would also need to be compelling reasons to include doses that were not tested in Phase II studies in the Phase III studies. Phase III studies are conducted in a much larger number of patients than in Phase II, and are designed to ‘confirm’ the results seen in Phase II in a sufficiently large patient population to satisfy the clinical trial sponsor and the regulatory authorities (…) (zie 2.9). Ook als de beweegreden voor een studie naar een bepaalde dosering gelegen kan zijn in wetenschappelijk inzicht ten behoeve van de gehele patiëntenpopulatie, en niet primair in een therapie voor de patiënten in de studie, zoals Van der Graaf in een latere verklaring heeft benadrukt, blijkt uit zijn verklaring wel dat een onderzoek in een Phase III studie naar een dosering die niet eerder is onderzocht ongebruikelijk is voor de vakman. De vakman zal er daarom een bijzondere beweegreden aan toedichten. De vakman leest in media release Novartis ook dat ‘participants will be equally randomized’ tussen de verschillende doseringen en placebo en ziet dus dat de 0.5 mg arm van de studie net zo groot is als de 1.25 mg arm. De vakman zal het onderzoek naar de geclaimde dosering in een Phase III studie om deze redenen zien als een pointer om juist die dosering te onderzoeken met een redelijke verwachting van succes. Als de vakman geen specifieke reden heeft om te betwijfelen of die dosering effectief is, zal hij/zij op basis van de informatie in media release Novartis met een redelijke verwachting van succes, die dosering onderzoeken. In zoverre oordeelt de voorzieningenrechter in lijn met de TKB (7.9 1e zinsdeel).
4.5.5.
Novartis betoogt echter dat de vakman wel een specifieke reden heeft om te betwijfelen of die dosering effectief is. De stand van de techniek zou hem/haar leren dat die dosering te laag is (‘teaching away’) waardoor de vakman meent dat die dosering niet zou werken. Novartis wijst daarbij op Webb (zie 2.5.6) in combinatie met Park 2003 (zie 2.5.5), Park 2005 (zie 2.5.7) en Kahan (2.5.4). Zij wijst daarbij met name op de volgende zin in Webb: “In dose response experiments, we found that a threshold of about 70% depletion of peripheral lymphocytes was required to see any efficacy, and thereafter, the dose response relationship between clinical benefit and lymphopenia was very steep.”
4.5.6.
Volgens Novartis en haar deskundigen zou deze passage in Webb er toe leiden dat de vakman zou betwijfelen of een dosering van 0.5 mg per dag (p.o.) een therapeutisch effect zou hebben, omdat op de prioriteitsdatum werd aangenomen dat de therapeutische werking van fingolimod bij RRMS werd bewerkstelligd door lymfocytensuppressie en hij/zij hieruit opmaakt dat daarvoor een drempelwaarde geldt van 70% reductie. De vakman zou dat combineren met de resultaten uit Park 2005 en Kahan, die openbaren dat er een lymfocytenreductie van 44% of 50% plaatsvond bij toediening van een dosering van 0.5 mg per dag (p.o.) aan transplantatie-patiënten. De vakman zou uit de combinatie van deze documenten concluderen dat de 0.5 mg per dag dosering onvoldoende is om de in Webb genoemde drempelwaarde te halen, aldus Novartis.
4.5.7.
Naar voorlopig oordeel is deze passage in Webb niet een zodanige ‘pointer away’, dat dat de succesverwachting van de vakman van de in media release Novartis aangekondigde Phase III studie met 0.5 mg per dag genoeg zou temperen. Zoals Mylan c.s. terecht stelt is Webb geen algemene vakkennis. Van een breed gedragen vooroordeel is voorshands niets gebleken.
4.5.8.
Maar ook als de vakman bekend zou zijn met Webb, zal hij/zij daar niet zoveel gewicht aan toekennen als Novartis nu doet. Webb betreft onderzoek gericht op transplantatie-patiënten, uitgevoerd bij muizen. De vakman zou mogelijk menen dat de studies bij transplantatie-patiënten relevant zijn voor de indicatie RRMS, zo blijkt uit Thomson (zie 2.5.8), maar het betreft geen klinische studie en het is niet specifiek op RRMS gericht zoals media release Novartis. Webb openbaart bovendien in dezelfde paragraaf dat er naast de werking van FTY720 op het lymfocyten-niveau, mogelijk ook andere mechanismen een rol spelen bij de therapeutische werking van fingolimod. Deskundige dr. C. Wolf merkt namens Mylan c.s. bovendien terecht op dat de 70% reductie waarover Webb schrijft een relatieve waarde is. Of de absolute waarden in de onderzochte patiëntenpopulatie vergelijkbaar is met die bij RRMS-patiënten, blijkt daar niet uit. Die waarden zouden bij transplantatie-patiënten hoger kunnen liggen, waardoor een sterkere reductie nodig is voor klinische effectiviteit. Volgens deskundige Wolfzou dit bijvoorbeeld veroorzaakt kunnen worden door infecties bij transplantatie-patiënten. Waarom de vakman zou menen dat de relatieve minimale lymfocytenreductie ook geldt als drempelwaarde voor klinische werking bij RRMS patiënten, en die zou relateren aan de resultaten van Park 2005 en Kahan, is niet duidelijk.
4.5.9.
De deskundigen van partijen lijken het erover eens dat het overzichtsartikel Thomson, dat gaat over toepassing van Fingolimod voor de indicatie RRMS, op de prioriteitsdatum de stand van de techniek voor EP 894 goed samenvat. Opmerkelijk is dat het artikel van Webb (en dus ook de door Novartis aangeduide drempelwaarde) in dit overzichts-artikel niet eens wordt genoemd. De vakman die desondanks bekend is met Webb zal zeker ook bekend zijn met Thomson en zien dat de drempelwaarde die Webb aanwijst, in Thomson niet wordt herhaald. Integendeel, in Thomson leest hij/zij: “In another study, the same dose [Vzr: 1 mg per dag] led to a 44% reduction in the number of blood lymphocytes in 32 subjects with or without hepatic impairment (Kovarik et al. 2005). A similar effect was also seen in a phase I study after single-dose administration of FTY720 (0.25-3.5 mg) to 20 stable renal transplant patients receiving a cyclosporine-based regimen (Budde et al. 2002). Although the higher doses of FTY720 produced a more rapid and sustained lymphocyte sequestration, the actual degree of this property was similar across the range of doses used in the study and no clear dose-response relationship was detected.” Daaruit zou hij/zij afleiden dat ook een lagere dosering tot een vergelijkbare lymfocytenreductie leidt, al duurt het langer om het resultaat te bereiken.
4.5.10.
Tot slot zou de vakman bij nauwkeurige lezing van Park 2005 zien dat de lymfocytenreductie van 70% in die studie ook niet is gehaald bij de dosering van 1.25 mg per dag (p.o.), zoals blijkt uit onderstaande figuur uit Park 2005, waarin Mylan c.s. met rode lijnen de lymfocytenreductie bij de 1.25 mg per dag dosering heeft weergegeven:
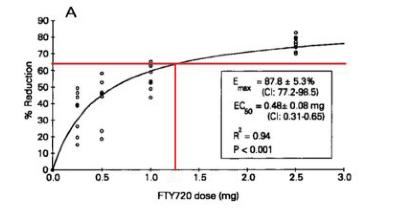
Uit media release Novartis weet de vakman echter dat de 1.25 mg per dag (p.o.) dosering therapeutisch zeer effectief is gebleken in een Phase II klinische studie voor de indicatie RRMS. Ook dat maakt dat hij weinig waarde zal toekennen aan de gegevens uit Webb.
4.5.11.
De vakman zou aan Webb naar voorlopig oordeel dus niet een zodanige contra-indicatie ontlenen dat hij/zij geen redelijke verwachting van succes meer zou hebben over de Phase III studie naar de geclaimde dosering van 0.5 mg per dag (p.o.).
4.5.12.
De voorgaande analyse geldt niet alleen voor media release Novartis, maar ook voor presentatie Novartis. Niet in geschil is dat presentatie Novartis alle kenmerken openbaart die in de hiervoor beschreven inventiviteitsanalyse ten opzichte van media release Novartis aan de orde kwamen. Daar komt echter nog bij dat de vakman in presentatie Novartis (zie 2.5.2) leest dat Novartis met de FDA heeft besproken om “even lower doses” te onderzoeken dan 0.5 mg per dag. Daaruit zal de vakman afleiden dat de FDA een succesverwachting had bij een dosering van 0.5 mg. Uit de Decision blijkt dat de TKB dit document niet bij haar beoordeling heeft betrokken. In een toekomstige oppositie- of nietigheidsprocedure zal dit document naar alle waarschijnlijkheid wel onderdeel uitmaken van de aangevoerde gronden.
4.5.13.
Ook Kappos (2.5.3) openbaart de hiervoor beschreven Phase III studie van Novartis met fingolimod in de doseringen 1.25 mg, 0.5 mg en placebo. Dit document zou de vakman combineren met media release Novartis of presentatie Novartis. Kappos openbaart daarover namelijk ook nog: ‘Ethical considerations relating to placebo use have been addressed by (…)’. Omdat dit alleen over placebo toediening gaat, zou de vakman daaruit opmaken dat die ethical considerations niet gelden voor de 0.5 mg dosering. Daarin zal hij de bevestiging zien dat van die dosering therapeutische werking mag worden verwacht.
4.5.14.
Novartis heeft nog gesteld dat er destijds een ziekenhuis zou zijn geweest, het Mount Sinaï ziekenhuis, dat weigerde deel te nemen aan de Phase III studie met de dosering van 0,5 mg per dag fingolimod, wegens bezorgdheid over de doeltreffendheid van deze dosering. Mylan c.s. heeft vervolgens echter gemotiveerd en onweersproken aangevoerd dat die weigering een andere studie betrof. Het Mount Sinaï ziekenhuis heeft wel meegewerkt aan de Phase III studie naar de 0,5 mg dosering.
4.5.15.
Interne documenten van Novartis waarin risico’s van de studie naar een dosering met 0.5 mg per dag zijn genoemd, kende de vakman niet en kunnen dus niet afdoen aan zijn/haar redelijke succesverwachting. Evenmin is van belang of de vakman de verwachting zou hebben dat de dosering van 0.5 mg per dag (p.o.) de dosering zou worden waarvoor een licentie zou worden verkregen. De vakman hoefde slechts de verwachting te hebben dat deze alternatieve dosering een effectieve behandeling van RRMS zou bieden.
4.5.16.
De motivering van de TKB in de Decision brengt de voorzieningenrechter niet tot een ander voorlopig oordeel. Zoals uit het voorgaande blijkt, is ook de TKB van oordeel dat media release Novartis schadelijk is voor de inventiviteit tenzij er in de stand van de techniek een ‘teaching away’ is (zie 2.8, overweging 7.9). De TKB is echter wel van oordeel dat Webb en de daarin geopenbaarde ‘drempelwaarde’ van 70% lymfocytenreductie ervoor zouden zorgen dat de vakman geen redelijke succesverwachting had (zie overweging 7.10 van de Decision). De TKB gaat ervan uit dat de vakman bekend is met Webb maar motiveert dat niet. Ook gaat de TKB ervan uit dat de vakman uit Webb zou afleiden dat er een drempelwaarde voor lymfocytenreductie is die eveneens geldt voor RRMS-patiënten. De TKB heeft blijkbaar niet de hiervoor aan de orde gekomen verweren daartegen bij de beoordeling betrokken. Daar wreekt zich mogelijk het feit dat de verleningsprocedure geen procedure op tegenspraak is, zodat de TKB niet of onvoldoende bekend was met die argumenten.
4.5.17.
De TKB heeft ook geen acht kunnen slaan op alle documenten die Mylan c.s. in het onderhavige kort geding heeft overgelegd en de daarbij behorende argumenten. De third party opinion van Mylan c.s. en presentatie Novartis (document TPO-D1 in die procedure) waarop Mylan c.s. zich nu beroept, had zij ook ingediend in de verleningsprocedure voor de TKB. Uit de Decision van de TKB blijkt dat de TKB die stukken niet bij de beoordeling heeft betrokken, omdat ze te laat in de procedure waren ingediend. Hetzelfde lijkt te gelden voor Kappos, dat ook niet genoemd wordt in de Decision. In hoeverre de argumenten die Mylan c.s. in dit kort geding naar voren heeft gebracht door derden voor de TKB zijn aangevoerd, is eveneens onduidelijk.
4.5.18.
Er is naar voorlopig oordeel dus een gerede kans dat de beslissing tot verlening van aanvraag EP 894 of in oppositie of, voor het Nederlandse deel van het octrooi, in een nietigheidsprocedure wordt vernietigd vanwege een gebrek aan inventiviteit. Voor het opleggen van een inbreukverbod in kort geding is om die reden geen grond. Of er ook een risico is dat het octrooi, indien wel inventief, nietig is vanwege onvoldoende plausibiliteit, hetgeen ook door Mylan c.s. is aangevoerd, kan dan ook in het midden blijven.
4.6.
In het tussenvonnis is al overwogen dat de provisionele voorziening niet toewijsbaar is (zie het tussenvonnis 3.1 onder A en 4.2). Ook is daarin al beslist dat de primaire vordering zal worden afgewezen (zie het tussenvonnis 3.1 onder B 1) en 2) en 4.14). De subsidiaire vorderingen delen nu dat lot.
Proceskosten
4.7.
Novartis zal als de overwegend in het ongelijk gestelde partij in deze procedure worden veroordeeld in de proceskosten. Mylan c.s. heeft begroting van de proceskosten voor 75% op de voet van artikel 1019h Rv gevorderd en voor 25% op basis van het liquidatietarief. Gelet op dit standpunt laat de voorzieningenrechter in het midden of de provisionele en primaire vorderingen van Novartis al dan niet zijn aan te merken als handhaving van intellectuele eigendomsrechten in de zin van artikel 1019 Rv. Mylan c.s. heeft opgegeven dat haar totale advocaat- en gemachtigde-kosten € 343.905 bedragen. De voorzieningenrechter merkt deze zaak aan als een complex kort geding in de zin van de indicatietarieven in octrooizaken van deze rechtbank, omdat er principiële juridische vragen aan de orde waren, twee zittingen nodig waren en nog aanvullende aktes zijn genomen na de eerste zitting. Daarvoor geldt een indicatietarief van 120.000,- en een liquidatietarief van € 1.524, waarbij de voorzieningenrechter 75% van de kosten toeschrijft aan de octrooi-grondslag en 25% aan de onrechtmatige daad-grondslag. De advocaat en gemachtigden-kosten worden derhalve begroot op € 90.000 voor de octrooirechtelijke grondslag en € 381 voor de onrechtmatige daad-grondslag, is samen € 90.381. Novartis zal daarom worden veroordeeld in de proceskosten ter hoogte van € 90.381, te vermeerderen met de volgende verschotten: € 676 griffierecht, € 17.200 deskundigenkosten en € 5.076 tolkenkosten, dus in totaal € 113.333.
5. De beslissing
De voorzieningenrechter
5.1.
wijst de vorderingen af;
5.2.
veroordeelt Novartis in de proceskosten, tot op heden aan de zijde van Mylan c.s. begroot op € 113.333;
5.3.
verklaart dit vonnis wat betreft de proceskostenveroordeling uitvoerbaar bij voorraad.
Dit vonnis is gewezen door mr. F.M. Bus en in het openbaar uitgesproken op 21 juni 2022.
Uitspraak 22‑03‑2022
Inhoudsindicatie
Kort geding. Voorlopig oordeel dat markttoetreding door generieke farmaceut voordat octrooi is verleend niet onrechtmatig is, ook niet als octrooi naar verwachting in de komende maanden verleend zal worden door EOB en octrooiconclusie al vaststaat.
Partij(en)
vonnis
RECHTBANK DEN HAAG
Team handel
zaaknummer / rolnummer: C/09/625743 / KG ZA 22-195
Vonnis in kort geding van 22 maart 2022
in de zaak van
de rechtspersoon naar vreemd recht
NOVARTIS AG,
te Bazel, Zwitserland,
eiseres,
advocaat mr. R.M. Kleemans te Amsterdam,
tegen
1. MYLAN B.V.,
te Amstelveen,
2. de rechtspersoon naar vreemd recht
MYLAN IRELAND LIMITED,
te Dublin, Ierland,
gedaagden,
advocaat mr. J.J.E. Bremer te Den Haag.
Partijen zullen hierna Novartis en Mylan c.s. genoemd worden en gedaagden ook afzonderlijk Mylan BV en Mylan Ltd. De zaak is voor Novartis inhoudelijk behandeld door mr. Kleemans voornoemd, mr. J.D. Drok en mr. A.F. Tadema, advocaten te Amsterdam en voor Mylan c.s. door mr. Bremer voornoemd, mr. M.H.J. van den Horst en mr. A.H. van Duijn, advocaten te Den Haag.
1. De procedure
1.1.
Het verloop van de procedure blijkt uit:
- -
de dagvaardingen van 1 maart 2022, met productie EP01 tot en met EP08;
- -
de akte houdende overlegging productie van Novartis, ingekomen ter griffie op 4 maart 2022, met productie EP09;
- -
de conclusie van antwoord, ingekomen ter griffie op 4 maart 2022, met productie GP01 tot en met GP12;
- -
de akte houdende overlegging producties van Novartis, ingekomen ter griffie op 8 maart 2022, met productie EP10 tot en met EP14;
- -
de akte overlegging producties van Mylan c.s., ingekomen ter griffie op 8 maart 2022, met productie GP13 tot en met GP15;
- -
de akte overlegging productie van Mylan c.s., ingekomen ter griffie op 8 maart 2022, met productie GP16 (proceskostenoverzicht);
- -
de akte overlegging productie van Novartis, ingekomen ter griffie op 8 maart 2022, met productie EP15 (proceskostenoverzicht);
- -
de pleitnotities van Novartis, ingekomen ter griffie op 9 maart 2022;
- -
de pleitnota van Mylan c.s., ingekomen ter griffie op 9 maart 2022;
- -
de akte overlegging productie van Mylan c.s., ingekomen ter griffie op 14 maart 2022, met productie GP16.1 (een geactualiseerd proceskostenoverzicht);
- -
de akte overlegging productie van Mylan c.s., ingekomen ter griffie op 14 maart 2022, met productie GP17;
- -
de mondelinge behandeling van 14 maart 2022 met aanwezigheid van partijen, advocaten, octrooigemachtigden en belangstellenden.
1.2.
Tijdens de mondelinge behandeling heeft Mylan c.s. toegezegd niet op de markt te komen met haar generieke fingolimod product (hierna in r.o. 2.5 nader omschreven), in het geval de bepaalde vonnisdatum van 22 maart 2022 niet gehaald wordt en het vonnis een aantal dagen later zal worden gewezen.
1.3.
Vonnis is bepaald op heden.
2. De feiten
2.1.
De Novartis-groep is een farmaceutisch concern, geleid door Novartis. De Novartis-groep ontwikkelt, produceert en verhandelt geneesmiddelen. Onder andere het geneesmiddel met de merknaam Gilenya®, met de werkzame stof fingolimod voor de behandeling van relapsing-remitting multiple sclerose. Gilenya is op 22 maart 2011 goedgekeurd in de Europese Unie door – kort gezegd – de verlening van handelsvergunningen. Aanvankelijk is Gilenya goedgekeurd voor gebruik bij volwassen patiënten, nadien ook voor pediatrische patiënten. De verlengde gegevensbeschermingsperiode voor Novartis voor het in de handel brengen van een generiek geneesmiddel waarbij Gilenya dient als referentiegeneesmiddel, loopt in onder meer Nederland af op 22 maart 2022.
2.2.
Novartis heeft op 16 juli 2015 een Europese octrooiaanvraag ingediend onder nummer EP 2 959 894 (hierna: aanvraag EP 894), getiteld “S1P-receptormodulatoren voor de behandeling van multiple sclerose”. Aanvraag EP 894 is een afgesplitste aanvraag, waarvan de grootmoederaanvraag (met nummer EP 2 037 906) op 25 juni 2007 is ingediend, die op haar beurt prioriteit inroept van GB0612721, gedateerd 27 juni 2006.
2.3.
De Examining Division van het EOBheeft de aanvraag EP 894 niet verleend op grond van niet-nieuwheid. Novartis is van deze beslissing in beroep gegaan bij de TKBvan het EOB. Tijdens de zitting van 8 februari 2022 heeft de TKB de beslissing van de Examining Division vernietigd en geoordeeld dat Novartis een octrooi toekomt. Het TKB heeft de zaak terugverwezen naar de Examining Division met de opdracht het octrooi te verlenen op basis van de enige conclusie van het hoofdverzoek. Deze conclusie luidt als volgt:
A S1P receptor modulator for use in the treatment of relapsing-remitting multiple sclerosis, at a daily dosage of 0.5 mg p.o., wherein said S1P receptor modulator is 2-amino-2-[2-(4-octylphenyl)ethyl]propane-1,3-diol in free form or in a pharmaceutically acceptable salt form.
2.4.
De Mylan-groep ontwikkelt, produceert en verhandelt onder meer generieke geneesmiddelen.
2.5.
Mylan Ltd heeft op 18 augustus 2021 een centrale handelsvergunning verkregen voor haar generieke fingolimod product, te weten een harde capsule van 0,5 mg, waarin de werkzame stof fingolimod is (hierna: fingolimod Mylan).
2.6.
Mylan BV is blijkens de bijsluiter van fingolimod Mylan de lokale Nederlandse vertegenwoordiger van Mylan Ltd voor fingolimod Mylan en verantwoordelijk voor het in de handel brengen in Nederland van dit product.
2.7.
Fingolimod Mylan is ingeschreven in de G-standaard voor maart 2022. Daarin is aangegeven dat fingolimod Mylan beschikbaar zal zijn vanaf 10 maart 2022.
2.8.
De advocaat van Novartis heeft Mylan c.s. op 16 februari 2022 een brief gestuurd met de sommatie om te bevestigen dat Mylan c.s. de bestaande en toekomstige rechten van Novartis ten opzichte van Gilenya zal respecteren en geen generieke fingolimod-producten op de markt zal brengen. Mylan c.s. heeft geen gehoor gegeven aan deze sommatie.
3. Het geschil
3.1.
Novartis vordert dat de voorzieningenrechter
A. bij wijze van provisionele voorziening:
i. indien en voor zover niet zeker is dat voor 22 maart 2022 vonnis wordt gewezen: Mylan c.s. direct of zo snel mogelijk na de mondelinge behandeling, voor zover nodig bij wege van mondelinge uitspraak, zal bevelen tot aan het in dezen te wijzen schriftelijke vonnis, zoals bedoeld onder B, zich in Nederland te onthouden van het vervaardigen, het in het verkeer brengen, verder verkopen, afleveren of anderszins verhandelen, dan wel voor een of ander aanbieden, in te voeren of in voorraad te hebben van een generiek fingolimod-product op de markt dan wel daarbij op onrechtmatige wijze betrokken te zijn; en
zal bepalen dat Mylan c.s. een onmiddellijk opeisbare dwangsom verbeurt van € 100.000,- per dag (bij aanbieden) dan wel ter keuze van Novartis, een onmiddellijk opeisbare dwangsom van € 1.000,- per product (bij uitleveren) dat zij in gebreke is ter zake van het onder i) te geven verbod; alsmede
bij vonnis, voor zover mogelijk uitvoerbaar bij voorraad:
primair:
1. Mylan c.s. zal verbieden om vanaf 22 maart 2022 tot aan het verlopen van EP 2 959 894 (hierna: EP 894) op 24 juni 2027 in Nederland inbreuk te maken op het Nederlandse deel van EP 894, en in het bijzonder fingolimod Mylan te vervaardigen, in het verkeer te brengen, verder te verkopen, af te leveren of anderszins te verhandelen, dan wel voor een of ander aan te bieden, in te voeren of in voorraad te hebben, dan wel daarbij op onrechtmatige wijze betrokken te zijn; en
2) zal bepalen dat Mylan c.s. een onmiddellijk opeisbare dwangsom verbeurt van € 100.000,- per dag (bij aanbieden) dan wel ter keuze van Novartis, een onmiddellijk opeisbare dwangsom van € 1.000,- per product (bij uitleveren) dat zij in gebreke is ter zake van het onder 1) te geven verbod;
subsidiair:
3) Mylan c.s. zal verbieden om vanaf de verlening van EP 894 tot aan het verlopen van EP 894 op 24 juni 2027 in Nederland inbreuk te maken op het Nederlandse deel van EP 894, en in het bijzonder fingolimod Mylan te vervaardigen, in het verkeer te brengen, verder te verkopen, af te leveren of anderszins te verhandelen, dan wel voor een of ander aan te bieden, in te voeren of in voorraad te hebben, dan wel daarbij op onrechtmatige wijze betrokken te zijn; en
4) Mylan c.s. zal bevelen om ieder product dat na de verlening van EP 894 inbreuk zal maken op het Nederlandse deel van EP 894 op eigen kosten in Nederland terug te halen; en
5) Mylan c.s. zal gebieden om alle afnemers van fingolimod Mylan uiterlijk bij de koop van fingolimod Mylan op een moment tussen 22 maart 2022 en het moment dat het octrooi wordt verleend, erover te informeren dat er binnenkort een octrooi zal worden verleend waarop fingolimod Mylan inbreuk maakt en dat dat betekent dat Mylan weliswaar op dit moment gerechtigd is om fingolimod Mylan te verkopen maar dat zij dat niet langer zal zijn op het moment dat het octrooi verleend wordt; en
6) Mylan c.s. zal gebieden dat zij op het moment dat zij door Novartis op de hoogte wordt gesteld dat EP 894 is verleend, onmiddellijk alle afnemers van fingolimod Mylan in Nederland schriftelijk bij brief informeert dat fingolimod Mylan niet langer verhandeld mag worden en door Mylan op eigen kosten zal worden teruggenomen;
7) Mylan c.s. zal gebieden om de verkoop van fingolimod Mylan aan alle afnemers nauwkeurig bij te houden zodat achteraf kan worden vastgesteld hoeveel van deze producten in Nederland zijn verkocht, en aan wie, en wat de verkoopprijs is geweest, inclusief eventuele kortingen, en wat de totale omzet is die daarmee is behaald, en welke winst daarmee is behaald, geverifieerd door een register accountant, en al deze informatie in ieder geval binnen dertig dagen nadat Novartis Mylan c.s. erover heeft geïnformeerd dat het octrooi is verleend, aan Novartis te verstrekken;
8) zal bepalen dat Mylan c.s. een onmiddellijk opeisbare dwangsom verbeurt van € 100.000,- per dag, dat zij in gebreke is ter zake van de onder 3) en 4) te geven verboden/bevelen, dat Mylan c.s. een onmiddellijk opeisbare dwangsom verbeurt van € 1.000,- per dag dat zij in gebreke is ter zake van de onder 5) en 6) te geven verboden/bevelen, en dat Mylan c.s. een onmiddellijk opeisbare dwangsom verbeurt van € 10.000,- per dag dat zij in gebreke is ter zake van het onder 7) te geven verbod/bevel;
alsmede zowel onder A en B, zowel primair als subsidiair:
I. de termijn voor het instellen van een eis in de hoofdzaak op grond van artikel 1019i Rvzal bepalen op zes maanden na het in dezen te wijzen vonnis; en
Mylan c.s. zal veroordelen in de volledige kosten van deze procedure conform artikel 1019h Rv.
3.2.
Ter onderbouwing van haar primaire vordering in 3.1 onder B 1) stelt Novartis – samengevat – dat Mylan c.s. onrechtmatig handelt door fingolimod Mylan na 22 maart 2022 op de markt te introduceren, terwijl zij weet dat naar verwachting ongeveer half juni 2022 octrooi zal worden verleend op aanvraag EP 894, waarna Mylan c.s. haar generieke product weer van de markt moet halen, omdat het valt onder de beschermingsomvang van de conclusie. Novartis heeft daarom een zeer spoedeisend belang bij haar primaire vordering.
3.3.
Volgens Novartis volgt de onrechtmatigheid van het handelen van Mylan c.s. uit het feit dat een octrooirecht materieel gezien werking heeft vanaf het moment dat de aanvraag is ingediend. Wanneer het octrooi is verleend, gaat het recht met terugwerkende kracht in tot aan het moment van indiening van de aanvraag. Om die reden kan worden beargumenteerd dat een octrooirecht al vanaf het moment van aanvraag daadwerkelijk bestaat en de aanvrager beschermt, aldus Novartis.
3.3.1.
Die systematiek blijkt onder andere uit artikel 33 TRIPs-Verdrag, waarin is opgenomen dat de minimale beschermingstermijn voor octrooien 20 jaar is “counted from the filing date”. Artikel 36 lid 6 ROW 1995bevestigt dit voor Nederlandse octrooien en artikel 49 lid 1 ROW 1995 voor Europese octrooien. Deze systematiek komt ook tot uitdrukking in artikel 71 en 72 ROW 1995, waarin is geregeld dat de aanvrager vóór de verlening van het octrooirecht recht heeft op een redelijke vergoeding van personen die (desbewust) gebruikmaken van de geoctrooieerde uitvinding. Als het octrooirecht pas door de verlening zou ontstaan, zou er geen rechtsbasis voor dit vergoedingsrecht zijn, aldus Novartis. In een normale situatie zou een octrooirecht voorafgaand aan de formele verlening niet worden gehandhaafd, met name om praktische redenen.
3.3.2.
De onderhavige situatie is volgens Novartis echter uitzonderlijk omdat door de beslissing van de TKB voorafgaand aan de formele verlening van het octrooi nu al vaststaat dat het octrooi (met terugwerkende kracht) toegekend zal worden en hoe de enige conclusie van het octrooi zal luiden. In deze situatie hoeft de redelijke vergoeding van artikel 71 en 72 ROW niet toegepast te worden, maar kan de dreigende inbreuk door Mylan c.s. op het te verlenen octrooi van Novartis worden verboden op grond van onrechtmatig handelen.
3.3.3.
De belangenafweging op basis waarvan deze beslissing kan worden genomen, is volgens Novartis door de wetgever al uitgevoerd in het kader van het wetsvoorstel dat heeft geleid tot de wet van 12 januari 1977, waarbij artikel 43A ROW 1910, de voorloper van artikel 71 ROW 1995, in de ROW 1910 is geïntroduceerd. Onder de ROW 1910 werden drie momenten onderscheiden, te weten de terinzagelegging van de octrooiaanvraag, de openbaarmaking van de octrooiaanvraag en de octrooiverlening. De openbaarmaking vond plaats indien de aanvraagafdeling, na onderzoek, van oordeel was dat de aanvraag zich voor octrooiverlening leende. In dit systeem was tussen de periode van terinzagelegging en openbaarmaking de (voornoemde) redelijke vergoeding verschuldigd. Voor de periode tussen openbaarmaking en octrooiverlening kon de octrooihouder op grond van onrechtmatig handelen schadevergoeding en winstafdracht vorderen van derden die inbreuk maakten op het openbaar gemaakte octrooi (artikel 44 lid 1 ROW 1910). De onderhavige situatie is volgens Novartis vergelijkbaar met de situatie die gold onder de ROW 1910 voor de periode tussen openbaarmaking en verlening van het octrooi, omdat door de TKB is besloten dat het octrooirecht op basis van aanvraag EP 894 zal worden verleend en dit octrooirecht dus definitief is vast komen te staan. De huidige artikelen 71 en 72 ROW 1995 moeten conform de duidelijke bedoelingen van de wetgever (zoals volgt uit de wetsgeschiedenis) in het onderhavige geval daarom zo worden uitgelegd dat wanneer Mylan c.s. na afloop van de marktexclusiviteit op 23 maart 2022 met fingolimod Mylan op de markt komt, dit onrechtmatig is tegenover Novartis, aldus nog steeds Novartis.
3.4.
Wanneer de door Mylan c.s. voorgenomen marktintroductie vanuit het hiervoor beschreven wetssystematisch en wetshistorisch perspectief al niet onrechtmatig zou zijn, dan moet de marktintroductie van fingolimod Mylan volgens Novartis alsnog onrechtmatig worden geacht, gezien de overige omstandigheden van dit geval. Wanneer Mylan c.s. fingolimod Mylan op de markt brengt, zal dit slechts voor een paar maanden zijn, totdat het octrooirecht op basis van aanvraag EP 894 daadwerkelijk wordt verleend. Dit leidt tot prijsbederf, welk prijsbederf (na verdwijning van het generieke product van de markt) meestal niet meer ongedaan gemaakt kan worden, zodat Novartis onherstelbare schade lijdt. Bovendien zou Novartis na octrooiverlening alle derde partijen die in het bezit zijn van fingolimod Mylan individueel moeten aanspreken om haar octrooirechten te kunnen handhaven. Daarnaast handelt Mylan c.s. in strijd met de belangen van derden (patiënten, apotheken), welke belangen ook moeten worden meegewogen.
3.5.
Aan haar subsidiaire (neven)vorderingen in 3.1 onder B 3) tot en met 8) legt Novartis – verkort weergegeven – ten grondslag dat nu al duidelijk is dat fingolimod Mylan inbreuk zal maken op het octrooi zodra aanvraag EP 894 is verleend. Omdat Mylan c.s. haar generieke fingolimod product in de G-standaard heeft laten opnemen en dreigt toe te treden tot de markt terwijl het moment van octrooiverlening van aanvraag EP 894 in juni 2022 verwacht wordt, heeft Novartis een spoedeisend belang bij een verbod op inbreuk op het te verlenen octrooi.
3.6.
Mylan c.s. voert gemotiveerd verweer. Zij betwist (kort samengevat) dat haar toetreding tot de markt voordat verlening van een octrooi op aanvraag EP 894 heeft plaatsgevonden een onrechtmatige daad kan zijn, dat het vaststaat dat en wanneer er octrooi verleend zal worden op aanvraag EP 894 en dat haar toetreding in de omstandigheden van dit geval onrechtmatig is. Zij betwist ook de geldigheid van het octrooi. Volgens Mylan c.s. zal aanvraag EP 894, als daarop al octrooi wordt verleend, in oppositie of (nationaal) in een nietigheidsprocedure worden herroepen/vernietigd omdat er sprake is van toegevoegde materie, een gebrek aan nawerkbaarheid (plausibiliteit), een gebrek aan nieuwheid en een gebrek aan inventiviteit. Om die reden is er van dreigende inbreuk ook geen sprake. Mylan c.s. bestrijdt op dezelfde gronden ook de spoedeisendheid van de vorderingen.
3.7.
Op de stellingen van partijen wordt hierna, voor zover van belang, nader ingegaan.
4. De beoordeling
Bevoegdheid
4.1.
De voorzieningenrechter is internationaal en relatief al bevoegd op grond van artikel 26 lid 1 Brussel I bis-Voomdat Mylan c.s. is verschenen en heeft verklaard ervoor te kiezen de internationale en relatieve bevoegdheid niet te betwisten.
Provisionele voorziening (3.1 onder A)
4.2.
Bij de provisionele vordering heeft Novartis geen belang meer gezien de toezegging van Mylan c.s. ter zitting (zie 1.2), zodat die vordering voor afwijzing gereed ligt.
Primair gevorderde verbod (3.1 onder B 1)
4.3.
De vraag die in dit kader beantwoord moet worden, is of sprake is van onrechtmatig handelen wanneer Mylan c.s. er na 22 maart 2022 (het einde van de marktexclusiviteit van Gilenya voor Novartis) voor kiest om op de markt te komen met fingolimod Mylan tot het moment dat het octrooi wordt verleend op basis van aanvraag EP 894.
4.4.
Met verwijzing naar de Guidelines for Examination van het EOB, Part E, Chapter XII, paragraaf 9.2, is zeer aannemelijk dat aanvraag EP 894 met de conclusie zoals door de TKB is geaccordeerd (vergelijk onder 2.3), verleend gaat worden. Mylan c.s. heeft gewezen op de mogelijkheid van intrekking of verdere afsplitsing door Novartis, maar de voorzieningenrechter acht die door Mylan c.s. genoemde mogelijkheden voorshands onvoldoende aannemelijk gemaakt. De huidige conclusie stelt Gilenya onder bescherming. Daarbij kan wel de tekst van de beschrijving nog aangepast worden. Tussen partijen is niet in geschil dat fingolimod Mylan voldoet aan alle kenmerken van de voornoemde vastgestelde conclusie. Dat de exacte inhoud van de beschrijving relevant is voor de vraag of fingolimod Mylan onder de beschermingsomvang van de conclusie van aanvraag EP 894 valt, is door Mylan c.s. niet gesteld en lijkt voorshands ook niet aan de orde. Naar voorlopig oordeel valt fingolimod Mylan dus onder de beschermingsomvang van een octrooi dat in de komende maanden naar verwachting door het EOB verleend zal worden op aanvraag EP 894.
4.5.
Vooropgesteld wordt dat op grond van artikel 49 lid 1 ROW 1995 een Europees octrooirecht kan worden uitgeoefend vanaf de datum van de publicatie van de verlening van het octrooi. Vanaf dat moment heeft de octrooihouder het uitsluitend recht de in de wet omschreven voorbehouden handelingen te verrichten (vergelijk artikel 53 ROW 1995). Nadat het octrooi is verleend, kan de octrooihouder met terugwerkende kracht voor de periode vanaf het moment van publicatie van de octrooiaanvraag, tegenover derden die handelingen hebben verricht die vallen onder artikel 53 ROW 1995, (onder omstandigheden) aanspraak maken op een redelijke vergoeding (vergelijk artikel 72 ROW 1995). Dat betekent dat in die periode volgens de ROW dus niet (met terugwerkende kracht) sprake is van aan de aanvrager en toekomstig octrooihouder voorbehouden handelingen. Daarmee resteert de vraag of de (voorgenomen) marktintroductie van fingolimod Mylan door Mylan c.s. in de onderhavige situatie wel onrechtmatig is volgens het gemene recht (artikel 6:162 BW), zoals Novartis stelt en Mylan c.s. betwist.
4.6.
Volgens Novartis is dit handelen ten eerste als onrechtmatig te kwalificeren omdat artikel 33 TRIPs-Verdrag verdragsstaten verplicht om gedurende 20 jaar octrooibescherming te bieden. In de onderhavige situatie waarin zeker is dat het octrooi verleend zal worden en waarbij de bewoordingen van de conclusie vaststaan, wordt die beschermingstermijn volgens Novartis te sterk uitgehold als zij niet nu al aanspraak kan maken op bescherming van haar materieel al toegekende recht. Omdat de ROW die bescherming niet biedt, impliceert de verplichting neergelegd in artikel 33 TRIPs-Verdrag dat Novartis bescherming kan ontlenen aan artikel 6:162 BW. Dit betoog wordt van de hand gewezen. Artikel 33 TRIPs-Verdrag dient volgens de literatuur zo uitgelegd te worden dat een systeem als het Nederlandse waarbij het octrooi niet met terugwerkende kracht 20 jaar bescherming biedt, daarmee verenigbaar is, zolang maar is voldaan aan de vereisten van artikel 62 lid 2 TRIPs-Verdrag, te weten een voldoende voortvarende aanvraagprocedure.Dat het vereiste van artikel 62 lid 2 TRIPs-Verdrag in deze zaak is geschonden, heeft Novartis niet gesteld. Dat verdragsrechtelijke verplichtingen ex artikel 33 TRIPs-Verdrag dwingen tot een oordeel dat handelingen die onder de nog niet verleende, maar al wel bekende, conclusies vallen, nu al onrechtmatig zijn jegens Novartis, is naar voorlopig oordeel daaruit dus niet af te leiden. De in de ROW bepaalde duur van de bescherming leidt ook niet tot een wetssystematische uitleg waarbij een (toekomstige) octrooihouder recht heeft op een materiële bescherming van 20 jaar. De voorzieningenrechter merkt daarbij op dat de lange termijn tussen de octrooiaanvraag en de beslissing tot verlening in deze zaak mede door Novartis zelf is veroorzaakt. Deze aanvraag betreft een afgesplitste (kleindochter) octrooiaanvraag, die pas op 16 juli 2015 is aangevraagd met een beroep op een prioriteitsdocument van 27 juni 2006 (vergelijk onder 2.2). Dat de effectieve beschermingsduur van het octrooi korter is dan 20 jaar, is mede daaraan te wijten.
4.7.
Ten tweede betoogt Novartis dat een octrooi materieel gezien al ontstaat vanaf het moment dat de aanvraag is ingediend. Zij wijst daarbij op het recht op een redelijke vergoeding op grond van artikel 71 en 72 ROW 1995, dat met terugwerkende kracht geldt na verlening. Als het octrooirecht materieel pas zou ontstaan bij verlening, zou er volgens Novartis geen basis zijn voor toekenning van de in die artikelen bepaalde vergoeding voor handelingen die plaatsvonden voor die verlening.
4.8.
In de eerste plaats merkt de voorzieningenrechter op dat een recht waaraan bij verlening terugwerkende kracht wordt toegekend, niet betekent dat er al voor die verlening een materieel recht bestond. Terugwerkende kracht betekent juist het tegendeel.
4.9.
Dat de Nederlandse wetgever handelingen die na de verlening van een Europees octrooi inbreuk maken, vanaf de beslissing tot verlening door het EOB als onrechtmatig heeft aangemerkt in de wetsgeschiedenis, kan ook niet worden aanvaard. De regeling in de artikelen 71 en 72 ROW 1995 zijn volgens Novartis overgenomen uit artikel 43A ROW 1910. De wetsgeschiedenis van dat artikel duidt erop dat de wetgever een handeling waarop de redelijke vergoeding zag, rechtmatig achtte tot het moment van de beslissing tot verlening door de Octrooiraad, maar vanaf het moment van die beslissing tot de verleningsdatum als een onrechtmatige daad aanmerkte. Uit die wetsgeschiedenis kan volgens Novartis afgeleid worden, dat het kantelmoment voor de vraag wanneer nog sprake is van een rechtmatige daad waarvoor een redelijke vergoeding verschuldigd is en wanneer dat handelen onrechtmatig wordt, de materiële verleningsbeslissing van de octrooiverlenende instantie is. Dit betoog volgt de voorzieningenrechter niet. Zoals Novartis zelf ook heeft onderkend in haar pleitnota, was het aanvraagsysteem van de ROW 1910 voor Nederlandse octrooien anders ingericht. De beslissing tot verlening van de Octrooiraad leidde tot een publicatie van het octrooi zoals onderzocht en beoordeeld door de Octrooiraad (‘openbaarmaking’), maar verlening vond pas plaats nadat de oppositietermijn was verstreken en er op een eventuele oppositie was beslist. In het tijdvak tussen die ‘openbaarmaking’ en de verlening bestond er recht op schadevergoeding (maar kon de houder van het openbaar gemaakte octrooirecht dat recht nog niet handhaven) en werd toepassing van het octrooi door derden gekwalificeerd als een onrechtmatige daad. Dat systeem is niet van toepassing bij Europese octrooien (en onder de ROW 1995 ook niet meer op Nederlandse octrooien). Pas na verlening van een Europees octrooi is oppositie mogelijk.
4.10.
Bij de implementatie van het EOVin de ROW 1910 in 1977-1978 heeft de wetgever dat verschil tussen Nederlandse octrooien onder de ROW 1910 en Europese octrooien ook onder ogen gezien. In de Memorie van Toelichtingbij die implementatie is – onder meer – het volgende opgenomen:
Onderdeel Y
(…)
Bij de octrooiverleningsprocedure langs Europese weg wordt de aanvrage eerst gepubliceerd, welke publikatie te vergelijken is met de terinzagelegging onder de Rijksoctrooiwet. Volgens het bij onderdeel AD in te voegen artikel 43B zal een gepubliceerde Europese aanvrage dan ook aanspraak kunnen geven op een zelfde redelijke vergoeding als een onder de Rijksoctrooiwet ter inzage gelegde aanvrage. Na toetsing van de Europese aanvrage door het Europees Octrooibureau volgt, anders dan onder de Rijksoctrooiwet, in voorkomend geval verlening van het Europees octrooi. Dit octrooi
biedt, zoals eerder vermeld, een in alle opzichten gelijke bescherming als een nationaal octrooi. Eerst na octrooiverlening kan oppositie worden ingesteld, die tot gehele of gedeeltelijke heropening van het octrooi kan leiden.
(…)
Onderdeel AB
(…)
In verband met de in het vierde en het vijfde lid van artikel 34 aan te brengen wijzigingen moge het volgende worden opgemerkt. De oppositieprocedure voor het Europees Octrooibureau vangt eerst aan na verlening van het Europees octrooi en eindigt in handhaving van het octrooi of wel gehele of gedeeltelijke herroeping ervan. Onder de Rijksoctrooiwet heeft oppositie plaats vóór de verlening van het octrooi; oppositie kan worden ingesteld na de openbaarmaking van de aanvrage om octrooi, dat wil zeggen nadat de aanvrage de toetsing door de Octrooiraad op formele en materiële vereisten heeft doorstaan. Een onder de Rijksoctrooiwet openbaar gemaakte aanvrage is daarom in vele opzichten te vergelijken met een Europees octrooi waartegen nog oppositie openstaat.
(…)
Onderdeel AE
(…)
Artikel 43A geeft de houder van een onder de Rijksoctrooiwet verleend octrooi het recht een redelijke vergoeding van derden te vragen voor handelingen die deze hebben verricht in de periode liggende tussen de volgens de Rijksoctrooiwet geschiede terinzagelegging van de octrooiaanvrage en de openbaarmaking daarvan. Voor de Europese aanvragen zijn de tijdstippen die de periode aangeven, waarbinnen de door derden verrichte handelingen moeten hebben plaatsgevonden, vervangen door voor die aanvragen vergelijkbare tijdstippen. Voor een Europees octrooi treedt de publikatie conform artikel 93 van het Europees Octrooiverdrag in de plaats van de terinzagelegging. Reeds eerder is vermeld dat een openbaar gemaakte Nederlandse aanvrage in vele opzichten is te vergelijken met een Europees octrooi, waartegen nog oppositie openstaat. Daarom is de officiële publikatie van de vermelding van de verlening van een Europees octrooi in de plaats getreden
van de openbaarmaking van een Nederlandse aanvrage. Vorenbedoelde afwijkingen zijn behalve in het eerste lid van artikel 43B ook verwerkt in het tweede en het vierde lid van dat artikel.
4.11.
Uit deze wetsgeschiedenis volgt dat de wetgever er in het systeem van de ROW 1910 vanuit is gegaan dat het moment van openbaarmaking van de octrooiaanvraag door de Octrooiraad gelijk gesteld kan worden aan het moment van publicatie van de octrooiverlening door het EOB. Dat is dus niet het moment waarop het EOB feitelijk de beslissing neemt om het octrooi te verlenen. Om die reden is een van artikel 43A ROW 1910 afwijkend artikel 43B toegevoegd aan de ROW 1910, specifiek voor Europese octrooien. Uit de wetsgeschiedenis van de artikelen 43A en 43B ROW 1910 kan daarom niet de conclusie worden getrokken dat de wetgever bedoeld heeft het toepassen van het octrooi in de periode tussen de feitelijke beslissing tot verlening door het EOB en de daadwerkelijke publicatie van verlening van een Europees octrooi als een onrechtmatige daad aan te merken. Dat dergelijke handelingen voor Nederlandse octrooien als onrechtmatig werden aangemerkt, is verklaarbaar vanuit de systematiek van octrooiverlening voor Nederlandse octrooien onder de ROW 1910. Overeenkomstige toepassing van de wetsgeschiedenis bij artikel 43A ROW 1910 op Europese octrooien is daarom niet aan de orde.
4.12.
De voorzieningenrechter is voorshands van oordeel dat er ook anderszins geen grond is om de handelwijze van Mylan c.s. als onrechtmatig aan te merken. Van de regeling voor een redelijke vergoeding in de ROW 1995 gaat naar voorlopig oordeel een negatieve reflexwerking uit. Dit recht op een vergoeding kent de wetgever met terugwerkende kracht toe. De wetgever heeft hier inderdaad gekozen voor een beperkter recht dan een handhavingsrecht, maar van een ‘lapmiddel’, zoals Novartis stelt, is daarmee nog geen sprake. Het moet immers gaan om een redelijke vergoeding. In de rechtspraak is in het verleden als redelijk aangemerkt de vergoeding die een redelijke licentienemer zou hebben willen betalen en waarmee een redelijk licentiegever bereid zou zijn geweest genoegen te nemen. In de weging van wat redelijk is, kunnen ook feitelijke omstandigheden worden meegenomen, zoals marktomstandigheden. Naar voorlopig oordeel zijn door Novartis genoemde risico’s als prijsdemping en ‘dumping’ inherent aan het recht van Mylan c.s. om, in de afwezigheid van een exclusief recht, de markt te betreden en kunnen zij niet als een vorm van oneerlijke mededinging worden aangemerkt. Zoals Mylan c.s. terecht opmerkt, is er in het tijdvak tussen de publicatie van de aanvraag en de verlening van een Europees octrooi geen leemte in de wet, maar is met de regeling van de redelijke vergoeding in artikel 72 ROW 1995 voor dit tijdvak sprake van een bewuste keuze van de wetgever. Uit het Doxyclycine-arrestvolgt voorshands dat er in dat geval geen ruimte is voor aanvullende werking van het gemene recht. Het door Novartis genoemde arrest Staat/Bondais ouder, zodat dat niet langer als de stand van de jurisprudentie gezien kan worden. Voor aanvullende werking van het gemene recht is derhalve geen plaats.
4.13.
Een zuivere belangenafweging, enkel gegrond op de wederzijdse belangen van partijen, kan – wat daar ook van zij – bij gebreke van onrechtmatig handelen door Mylan c.s. niet tot een ander oordeel leiden.
4.14.
Het voorgaande betekent dat de primaire verbodsvordering genoemd in 3.1 onder B 1), evenals de daarop gegronde nevenvordering genoemd in 3.1 onder B 2), bij eindvonnis in dit kort geding zal worden afgewezen.
Subsidiaire vorderingen
4.15.
Novartis heeft ook een spoedeisend belang bij haar subsidiaire vorderingen genoemd in 3.1 onder B 3) tot en met B 8). Publicatie van de octrooiverlening op aanvraag EP 894 zal mogelijk begin juni plaatsvinden en waarschijnlijk in de periode tussen begin juni en begin augustus 2022. Mylan c.s. heeft een aantal situaties genoemd die mogelijk tot vertraging van de publicatie kan leiden, zoals het indienen van opmerkingen door belanghebbende partijen naar aanleiding van wijziging van de beschrijving. Er is echter geen sprake van dat die situaties zeker zullen voorvallen en tot grote vertraging zullen leiden. Mylan c.s. is voornemens om op de markt te komen en heeft duidelijk gemaakt dat zij niet voornemens is daarmee te stoppen op het moment van verlening, omdat zij het octrooi niet geldig acht.
4.16.
Mylan c.s. heeft tegen de subsidiaire vorderingen nietigheidsverweren gevoerd (zie 3.6). Zij wijst erop dat zij in haar verdediging is geschaad doordat zij haar verweer met de hoogste spoed in slechts een paar dagen kon voorbereiden. Met name was zij daardoor niet in de gelegenheid haar stellingen over de nietigheid van aanvraag EP 894 te onderbouwen met bewijsstukken zoals deskundigenverklaringen, aldus Mylan c.s.
4.17.
Dit verweer slaagt. De grote spoed waarmee dit kort geding is ingepland en de daarbij gehanteerde termijn voor een conclusie van antwoord die Mylan c.s. wenste te nemen, hing samen met de primaire vordering. De subsidiair gevorderde verbodsvoorziening (de vordering in 3.1 onder B 3)) is gericht op de periode vanaf de publicatie van de verlening en zal dus niet eerder dan in juni 2022 van belang worden voor Novartis. Dat geldt ook voor de vordering in 3.1 onder B 4), B 6) en B 7). Die laatste vordering ziet weliswaar ook op het bijhouden van gegevens in de periode tot publicatie van de octrooiverlening, maar met name op het opgeven van die gegevens aan Novartis na de verlening. De voorzieningenrechter gaat er voorshands van uit dat Mylan c.s. een zodanige administratie voert dat opgave achteraf ook mogelijk is zonder een bevel van de voorzieningenrechter om die gegevens te bewaren.
4.18.
Voor al deze vorderingen geldt dat er nog voldoende tijd is om Mylan c.s. meer tijd te gunnen voor wederhoor, zonder dat dit de belangen van Novartis ernstig schaadt. Mylan c.s. zal daartoe dus in de gelegenheid worden gesteld.
4.19.
De vordering in 3.1 onder B 5) is ook gericht op een voorziening in de periode tot de publicatie van de verlening. Een afweging van de wederzijdse belangen brengt mee dat ook voor de beoordeling van die vordering wordt gewacht op het voorlopig oordeel dat de voorzieningenrechter zal geven over de geldigheid van aanvraag EP 894 nadat partijen in de gelegenheid zijn geweest zich daarover nog uit te laten. Ook hier geldt dat in een kort geding waarin alleen de subsidiaire vorderingen aan de orde waren, Novartis die beoordeling had moeten afwachten.
4.20.
Mylan c.s. wordt in de gelegenheid gesteld haar nietigheidsverweren verder te substantiëren door daarover uiterlijk dinsdag 12 april 2022 een akte te nemen. Novartis mag daarop reageren bij akte van 2 mei 2022. Daarna zal een (voortzettings-)zitting plaatsvinden op dinsdag 17 mei 2022 om 15:00 uur, waarin partijen ieder 30 minuten (eerste termijn, aan de hand van pleitnotities) en 10 minuten (re- en dupliek) hun stellingen die zien op de nietigheidsverweren van Mylan c.s. kunnen bepleiten. Vervolgens zal de voorzieningenrechter beslissen op de subsidiaire vorderingen en de proceskosten.
5. De beslissing
De voorzieningenrechter
5.1.
bepaalt dat Mylan c.s. uiterlijk op 12 april 2022 om 10:00 uur een akte mag nemen als omschreven in r.o. 4.20,
5.2.
bepaalt dat Novartis uiterlijk op 2 mei 2022 om 10:00 uur een antwoordakte mag nemen,
5.3.
bepaalt dat de mondelinge behandeling van het kort geding wordt voortgezet op dinsdag 17 mei 2022 om 15:00 uur,
5.4.
houdt iedere verdere beslissing aan.
Dit vonnis is gewezen door mr. F.M. Bus en in het openbaar uitgesproken op 22 maart 2022.